12 GMO-legemidler i klinisk utprøving til mennesker og dyr
12.1 Innledning
I dette kapitlet beskrives problemstillinger, veivalg og utvalgets tilrådninger for vurderinger av GMO-legemidler som skal brukes i klinisk utprøving av legemidler til mennesker og dyr. Mandatpunket omhandler å vurdere norsk praksis for vurdering og godkjenning av GMO-legemiddel til klinisk utprøving, sammenligne med EUs praksis, og eventuelt foreslå endringer. Kapitlet tar blant annet for seg relevante prosesser i Norge og EU, historikk og norsk forvaltningspraksis på området, og legger fram en sammenligning med praksis i EU. Dette inkluderer hvilke utfordringer søkere har opplevd, og hvilke endringer som har blitt gjort i forvaltningen på dette området i Norge, inkludert endringer som er foretatt i løpet av utvalgsperioden. Videre omtales klinisk utprøving av GMO-legemidler til dyr i et eget underkapittel, men mesteparten av dette kapitlet omhandler klinisk utprøving til mennesker.
Det beskrives en vurdering av alternative veivalg, og forslag til endringer av regelverk og vedtaksmyndighet. Utvalget har vært samstemt i mange av vurderingene for klinisk utprøving av GMO-legemidler til mennesker. Utvalget er enig i at vedtaksmyndighet for klinisk utprøving av GMO-legemidler til mennesker bør være Legemiddelverket, og foreslår samlet flere forenklinger. Utvalgets vurderinger her samsvarer stort sett med nåværende regulering, som har blitt endret i løpet av utvalgsperioden. Utvalget deler seg i vurderinger som går på endringer utover den nåværende reguleringen, blant annet i spørsmål om høring og visse aspekter av regulering og forvaltning av klinisk utprøving av GMO-legemidler til dyr, og i hvilket lovverk reguleringen skal ligge. Anbefalingene beskrives gjennom kapitlet og er også oppsummert til slutt i kapitlet i 12.7 og 12.8.
For mer detaljert informasjon om gjeldende og tilstøtende regelverk for GMO-legemidler vises det til kapittel 6.
12.1.1 Overordnet om GMO-legemidler i Norge og EU
Norge er gjennom EØS-avtalen knyttet opp mot legemiddelregelverket og GMO-regelverket i EU, som sammen utgjør reguleringen av legemidler som består av eller inneholder genmodifiserte organismer (GMO-legemidler). Norsk regulering og forvaltningspraksis er i stor grad den samme som i resten av EØS-området. GMO-legemidler til klinisk utprøving godkjennes etter nasjonalt GMO-regelverk, og krever godkjenning etter legemiddelloven, helseforskningsloven og genteknologiloven i Norge. Regelverk og beskrivelse av forvaltningen på området er omtalt i kapittel 6. I løpet av årene 2018 til og med 2021 har forvaltningen mottatt seks søknader om klinisk utprøving av GMO-legemidler til mennesker, mens det ikke er kommet inn søknader om utprøving av GMO-legemidler til dyr. En observasjon i etterkant er at søknadsbehandlingen etter de regelverk som kommer til anvendelse ved klinisk utprøving av GMO-legemidler, i starten, kan ha vært utfordrende å koordinere for myndighetene. Saksbehandlingen tok i flere saker lengre tid enn det som anses hensiktsmessig med mulige negative konsekvenser for pasienters kliniske behandlingstilbud i aktuelle studier. Dette fikk uheldige konsekvenser for oppstart av enkelte studier. Ifølge Dagens Medisin var en av studiene i ferd med å bli trukket fra Norge grunnet krav knyttet til saksbehandlingen.1
Genmodifiserte organismer i legemidler er som regel genmodifiserte mikroorganismer (GMM), som virus, bakterier eller celler som har fått innsatt, endret eller fjernet genetisk materiale. I dag foregår en rask utvikling på området, og det er forventet at stadig flere slike GMO-legemidler vil forskes frem ettersom teknologiene utvikles og kunnskapen om gener og sykdommer øker. GMO-legemidler til mennesker er i hovedsak utviklet til såkalt avansert terapi, som genterapi for å korrigere følger av genforandringer betinget i sjeldne arvelig sykdommer, og som celleterapi i kreftbehandling. Til dyr er det i hovedsak genmodifiserte virusvaksiner som er utviklet og markedsføres, men også til mennesker går utviklingen i retning av genmodifiserte virusvaksiner. Eksempler på dette er GMO-vaksiner mot ebola, og GMO-vaksinene fra AstraZeneca og Johnson & Johnson mot covid-19. Nye teknikker som CRISPR/Cas9 forventes også å få stor betydning i utviklingen av slike legemidler fremover. I 2023 ble den første CRISPR-baserte genterapien, til behandling av blodsykdommene sigdcelleanemi og beta-thalassemi, søkt godkjent i Europa og USA. En rekke tilsvarende legemidler er for tiden i klinisk utprøving.
En klinisk utprøving gjennomføres blant annet for å finne ut hvordan et legemiddel virker, hvilke bivirkninger det har, og hvordan det omsettes i kroppen. Begrepet klinisk utprøving brukes om forskning etter gjennomført prekliniske dyreforsøk både når utprøvingen skjer i et menneske eller i en dyreart, og uavhengig av om legemiddelet er til mennesker (humanmedisin) eller dyr (veterinærmedisin).
Området GMO-legemidler bringer sammen to ulike regelverk med ulike formål, og to forvaltningssektorer som i utgangspunktet har ulike målsetninger. Der hvor legemiddelsektoren har som hovedformål å sikre god og sikker behandling med legemidler, har miljømyndighetene som formål å trygge samfunn og miljø mot skade fra menneskelige aktiviteter. Samtidig har genteknologiloven som formål å sikre at genteknologi benyttes til samfunnets beste, og skal ikke være et hinder for innovasjon og utvikling av GMO-produkter som kan komme samfunnet til gode, uten negative følger for helse, miljø, bærekraft eller etikk. Hvordan man velger å forene disse ulike målsetningene, hvor legemiddelregelverk har pasienten i fokus, mens GMO-regelverk vektlegger samfunn og miljø i stort, kan være forskjellig i ulike land. Norge skiller seg fra øvrige EU/EØS-land ved å ha spesifikke krav etter genteknologiloven om vurdering av bærekraft, samfunnsnytte og etikk, i tillegg til kravene om vurdering av helse- og miljørisiko2,3.
Godkjenning av GMO i klinisk utprøving etter GMO-regelverket i EU skjer av nasjonale myndigheter, og praksis for GMO-godkjenningen er forskjellig fra land til land. Det er tre hovedmodeller for godkjenning av klinisk utprøving av GMO-legemidler til mennesker; godkjenning som innesluttet bruk (tre land), godkjenning som utsetting i forskningsøyemed (elleve land), eller en kombinasjon av innesluttet bruk og utsetting (ti land). Norge har sistnevnte modell, der praksis siden 2018 har vært at pasienten mottar GMO-legemidlet i en fasilitet godkjent for innesluttet bruk, og at utprøvingen i tillegg har en tillatelse til utsetting, som dekker eventuell utskilling av GMO etter at pasienten har forlatt innesluttet-fasiliteten. EU-kommisjonen har gitt visse føringer i sitt forslag til ny legemiddelregulering (april 2023, omtalt lenger ned) for hvordan vurdering og godkjenning av GMO-legemidler i kliniske utprøvinger med mennesker skal skje. Det vil samtidig være et nasjonalt handlingsrom til å bestemme norsk praksis på området når studier skal vurderes spesifikt i Norge, eller når Norge er koordinerende land i multisenterstudier. Det er følgelig et handlingsrom for å bestemme norsk praksis på området.
I figur 12.1 under vises en skjematisk oversikt over saksgangen for søknader og saksbehandling for kliniske utprøvinger med GMO-legemidler. Den første viser for utprøving til mennesker og den andre for utprøving til dyr.

Figur 12.1 Flytdiagram som viser dagens ordning for vurdering av søknader om klinisk utprøving med GMO-legemidler. REK-KULMU er Regional komité for medisinsk og helsefaglig forskningsetikk ved komiteene for klinisk utprøving av legemidler og medisinsk utstyr.
12.1.2 Utvalgets avgrensninger og skillelinjer
Mandatpunket angående kliniske utprøvinger er å Vurdere norsk praksis for vurdering og godkjenning av GMO-legemiddel til klinisk utprøving, samanlikne med EU sin praksis, eventuelt foreslå endringar. Utvalgets vurdering og forslag til endringer angår derfor kun klinisk utprøving av GMO-legemidler til mennesker og dyr, og ikke markedsføringstillatelse for GMO-legemidler.
Utvalget har valgt å skille mellom klinisk utprøving av GMO-legemidler til mennesker og dyr. Dette anses hensiktsmessig siden det er eget regelverk for hver av disse legemiddelkategoriene, og fordi handlingsplaner i EU4 og i Norge5 angående legemiddelutprøving bare omhandler klinisk utprøving av legemidler til mennesker. Ved utprøving av legemidler til dyr er risikoen og potensialet for eksponering i miljøet ofte større enn ved utprøving av legemidler til mennesker. Spesielt gjelder dette ved klinisk utprøving på dyrearter der vi har nærstående, viltlevende arter og/eller populasjoner. Myndighetene skal også vurdere risiko for de personer som håndterer legemidlet samt konsumentrisiko, herunder tilbakeholdelsestider, ved utprøving av legemidler til dyr. Forskjellene i regelverk og vurderingsgrunnlag gjør at det er enklere å diskutere praksis og alternative løsninger ved å skille på legemidler til mennesker og legemidler til dyr. Norsk forvaltning har så langt ikke mottatt noen søknader om klinisk utprøving av GMO-legemidler til dyr, og man har derfor begrenset erfaring på området. Kapittel 12.5 angår dyr spesielt.
12.1.3 Om relevante EU-prosesser på området
Klinisk utprøving, også kalt kliniske studier, er et komplekst område som berøres av flere regelverk. Legemiddelutviklere peker på flere forhold som er viktige for at de skal kunne gjennomføre kliniske studier, herunder tilgang til pasienter og egnede studiesteder, men også regulatoriske forhold. Dette er ikke en ny problemstilling, og EU iverksatte tilbake i 2016 derfor en handlingsplan for kliniske studier med legemidler til avansert terapi. Fokus var å tydeliggjøre og forenkle søknadsprosesser for kliniske studier til mennesker, i tråd med et uttalt politisk mål om å få flere kliniske studier til EU.
I sammenheng med dette arbeidet initierte EU-kommisjonen i desember 2016 et arbeid for å harmonisere praksis for GMO-legemidler på tvers av landene, det såkalte GMO-interplay arbeidet. Fokus for arbeidet har vært utvikling av felles veiledere og søknadsskjema for grupper av GMO-legemidler. Norske myndigheter ved Legemiddelverket og Miljødirektoratet har deltatt i disse prosessene. Arbeidet har også vært nyttig for avklaring av EU/EØS-regelverket om GMO-legemidler opp mot praksis i Norge.
Etter hvert ble handlingsplanen om avansert terapi fra 2016 fulgt opp av en egen legemiddelstrategi (fra desember 2020), blant annet med formål om å legge bedre til rette for den teknologiske utviklingen som skjer på legemiddelområdet. Om legemidler som består av eller inneholder GMO skriver EU-kommisjonen:
The regulatory requirements for the authorisation of medicines for human use that contain or consist of genetically modified organisms (GMOs) should be fit for purpose when it comes to addressing the specificities of medicines and the conduct of clinical trials with those products in the EU (which is currently hindered by the fragmentation of national requirements). Solutions will be explored during the evaluation of the pharmaceutical legislation. In general, consideration should be given to mechanisms for the continuous and timely adaptation of its technical requirements in light of emerging science and technologies with a view to enhance effectiveness to protect human health whilst minimising harmful impacts on the environment.
GMO er regulert etter eget EU-regelverk, som har som formål å hindre skade på helse og miljø av GMO-en. EU har omtalt erfaring med GMO-reguleringen i forbindelse med det ekstraordinære og midlertidige unntaket for GMO-godkjenning i kliniske utprøvinger av covid-19 vaksiner. Europaparlamentet og rådet skriver i sin begrunnelse for unntaket at: erfaring viser at for kliniske studier med GMO-legemidler er prosessen knyttet til miljørisikovurderingen kompleks og kan ta svært lang tid.
EU-kommisjonen har også utarbeidet en strategi og iverksatt tiltak for å begrense legemidlers tilførsel til miljøet, uavhengig av om legemidlet inneholder GMO, på grunn av mulige negative effekter på blant annet dyrs utvikling og reproduksjon, jamfør EU-kommisjonens strategi om legemidler i miljøet fra 20196. Det er i forbindelse med denne blant annet utviklet nye veiledninger for miljørisikovurdering av legemidler til markedsføring. Denne strategien er en del av EUs grønne giv (EU Green Deal), sammen med blant andre sirkulær økonomistrategien, biodiversitetsstrategien og matstrategien (farm to fork). GMO-legemidler er ikke spesifikt nevnt i disse strategiene, samtidig er GMO regulert etter eget EU-regelverk, som har som formål å hindre skade på helse og miljø av GMO-en.
Den 23. april 2023 publiserte EU-kommisjonen sine forslag til regelverksendringer på legemiddelområdet, herunder også GMO-legemidler i klinisk utprøving til mennesker7. De reglene som blir vedtatt, vil også gjelde i Norge, i tillegg til det som er nasjonale regler. Utvalget har ikke hatt tid til å diskutere disse anbefalingene i fellesskap. Flertallet vil imidlertid synliggjøre overlapp mellom egne anbefalinger (som utdypes senere i dette kapittelet) og EU-kommisjonens forslag samt legge til noen generelle vurderinger av de øvrige forslagene. Forslagene i den nye forordningen gjelder ikke for legemidler til dyr (jamfør fortalen i forslag til forordning), og det som er beskrevet under, gjelder derfor for legemidler til mennesker:
EU-kommisjonen anbefaler en forenklet godkjenning for kliniske studier med GMO-legemidler som kvalifiserer for skjemaer med en forhåndsutfylt miljørisikovurdering. Det samme anbefaler et samlet utvalg i denne NOU-en.
EU-kommisjonen foreslår å unnta kliniske studier med GMO-legemidler fra flere av kravene i utsettingsdirektivet, direktiv 2001/18/EF, spesifikt artikkel 6-11. Dette innebærer blant annet unntak fra krav om høring, slik utvalgsflertallet også anbefaler.
Kommisjonen anbefaler videre at bestemmelsene knyttet til godkjenning/miljørisikovurdering av GMO-legemidler i klinisk utprøving legges til forordning (EU) nr. 536/2014 om kliniske utprøvninger av legemidler til mennesker gjennom bestemmelser som utfyller eksisterende lovgivning (delegated acts). Utvalgsflertallet anbefaler også at regler for GMO-legemidler i Norge flyttes til helserelevant regelverk.
EU-kommisjonen foreslår at søknad og risikovurdering og godkjenning av kliniske utprøving etter GMO-regelverket skal foregå via CTIS (den felles EU-portalen for kliniske studier), slik utvalgsflertallet også anbefaler.
EU-kommisjonen anbefaler videre at GMO-godkjenningsprosessen for studier koordineres fra ett land, som innebærer at det ikke gjøres individuelle godkjenninger i enkeltland ved multisenterstudier. Vurderingen skal baseres på en vitenskapelig vurdering fra CHMP (komité under de europeiske legemiddelmyndighetene (EMA)) som sendes til myndigheten i landet som koordinerer prosessen. Flertallet støtter disse anbefalingene og viser til at en av de største barrierene for kliniske studier med GMO-legemidler i Europa er fragmenterte godkjenningsprosesser. Flertallet mener det er svært viktig at Norge, i tilfellene der Norge er koordinerende land, etterstreber en effektiv godkjenningsprosess under ledelse av Legemiddelverket slik utvalget foreslår.
EU-kommisjonen foreslår også at det i hastetilfeller gis midlertidig unntak fra krav om miljørisikovurdering etter GMO-regelverket for legemidler som ikke enda har markedsføringstillatelse, herunder til folkehelsetiltak og behandling av enkeltpasienter med stort behov (Compassionate use). Flertallet støtter også dette.
Flertallet anmoder norske myndigheter om å ta en aktiv pådriverrolle i den politiske oppfølgingen og implementeringen av de foreslåtte endringene i EU/EØS parallelt med endringer i det norske regelverket.
12.1.4 Nasjonale prosesser
I Norge fikk man også en nasjonal handlingsplan for kliniske studier på det humanmedisinske området i januar 2021, med målsetning om å doble antallet kliniske studier innen 2025. Utfordringsbildet er sammensatt, og handlingsplanen berører derfor flere aktører, deriblant sykehusene, forvaltningen, utdanningsinstitusjonene, primærhelsetjenesten, legemiddelindustri og ikke-kommersielle forskningsmiljø. Det anses som at klinisk utprøving generelt kommer pasientene til gode. Når det gjelder godkjenning av utprøving av GMO-legemidler er det i handlingsplanen nevnt at Regjeringen ønsker å legge til rette for gode prosesser og rask saksbehandling av kliniske studier med slike legemidler.
Utfordringer med gjeldende regelverk for GMO-legemidler har vært diskutert over tid i Norge, og i 2020 gjennomførte Klima- og miljødepartementet en offentlig høring av forslag om endring av genteknologiloven. Ett av forslagene var at genteknologilovens krav om vurdering av bærekraft, samfunnsnytte og etikk, i tillegg til de vurderinger av samfunnsnytte og etikk som gjøres av Regionale komiteer for medisinsk og helsefaglig forskningsetikk (REK) etter legemiddelregelverk og helseforskingsloven, ikke lenger skulle gjelde for utsetting av GMO i legemidler til klinisk utprøving. Dette var begrunnet i at disse kriteriene i stor grad, med unntak fra bærekrafts-kriteriet, også ble vurdert i forbindelse med godkjenning av klinisk utprøving etter legemiddelregelverk.
Høringsinstansene var delt i sitt syn. De som støttet forslaget, la i hovedsak vekt på at kravet om vurdering av disse kriteriene er overflødig, spesielt innen humanmedisin, og at unntaket vil gjøre søknadsprosessen enklere. Noen høringsinstanser gikk imot forslaget, enten i sin helhet eller delvis. Flere uttalte at lovforslaget ikke burde omhandle veterinærmedisin. For veterinærmedisinske legemidler ble det særlig vist til at aspektene knyttet til samfunnsnytte, bærekraft og etikk ikke er godt nok dekket av annet regelverk enn genteknologiloven. Lovforslaget er fremdeles under arbeid. Nærmere informasjon om høringen og høringsuttalelsene finnes på høringssiden til endringsforslaget8. Mange av høringssvarene pekte også på behov for større regelverksendringer for GMO-legemidler, blant annet knyttet til risikovurderinger.
Utvalgets vurderinger blir altså til i en tid med endringsprosesser. Etter gjeldende regelverk, faller GMO-legemidler, også de som er utviklet med genredigering, under GMO-regelverket. GMOer, inkludert genredigerte virus, bakterier og celler til bruk som legemidler, vil kreve tillatelse etter GMO-regelverket i EU/EØS, i tillegg til legemiddelregelverket, og endringer i regulering for andre GMO-bruksområder kan ha betydning for framtidig regulering av GMO-legemidler.
12.2 Nærmere om historikk og norsk praksis for godkjenning av klinisk utprøving av GMO-legemidler til mennesker etter genteknologiloven
I perioden 2018-2021 behandlet myndighetene seks søknader om utsetting av GMO til klinisk utprøving i mennesker. Saksbehandlingstiden i alle etater ble redusert for siste søknad mottatt i 2021, sammenlignet med første søknad mottatt i 2018. I det følgende belyses historikk og utvikling av praksis i Norge.
12.2.1 Samhandling og praksis om godkjenning av GMO til legemiddelutprøving før 2018
Antallet søknader om klinisk utprøving av GMO-legemidler til mennesker i Norge var marginalt. Det ble ikke søkt om studier med GMO-legemidler til dyr. Helsedirektoratet og Miljødirektoratet vurderte om studien innebar innesluttet bruk eller utsetting basert på studieoppsettet.
Helsedirektoratet godkjente som hovedregel studien som innesluttet bruk. Det var en forståelse mellom etatene at dette var det enkleste for søker og vurdert som forsvarlig med tanke på studiedesign. Miljødirektoratet hadde ikke mottatt eller godkjent en klinisk studie med GMO-legemiddel som utsetting før 2018.
12.2.2 Samhandling og praksis om godkjenning av GMO til legemiddelutprøving 2018 – 2021
12.2.2.1 Avklaringer i EU/EØS
I desember 2016 tok EU-kommisjonen initiativ til å opprette ekspertgrupper – GMO interplay – som skulle se på samspillet mellom GMO-regelverket og legemiddelregelverket for kliniske studier. Det var totalt fire grupper9, og det ble nominert fageksperter fra hvert land og fra begge regelverk. Miljødirektoratet og Legemiddelverket har vært representert inn i gruppen GMO-pharma. Helsedirektoratet har blitt konsultert i saker som omfatter innesluttet bruk. GMO-pharmagruppen møttes første gang i februar 2017, og de første møtene handlet om en felles forståelse for hva som faller inn under GMO-regelverket (GMO-definisjoner) ved klinisk utprøving av legemidler som inneholder GMO.
12.2.2.2 Samarbeidsmøter mellom etatene
I løpet av våren 2018 var flere aktører i kontakt med ulike myndigheter om godkjenning av kliniske studier med GMO-legemidler. Som forberedelse til behandling av disse søknadene ble det arrangert et samarbeidsmøte mellom etatene Helsedirektoratet, Miljødirektoratet, Legemiddelverket, samt Regionale komiteer for medisinsk og helsefaglig forskningsetikk (REK) i mai 2018.
Formålet med samarbeidsmøtene var å få klarhet i gjeldende regelverk på området og saksbehandlingstider etter hvert regelverk. Det var enighet om at det var viktig å sikre at etatene hadde omforent informasjon til søker om regelverket på området. Ved mottak av søknad ville etatene orientere hverandre, og henvise søkere til de rette etater for å sikre at søker har de godkjenninger som behøves før oppstart av studien.
12.2.2.3 Oppdrag til VKM om helse- og miljørisikovurdering
Vitenskapskomitéen for mat og miljø (VKM) fikk fra høsten 2017 oppdrag om risikovurdering av GMO i legemidler fra Miljødirektoratet. Det ble ansett som hensiktsmessig at vurdering av risiko og håndtering av risiko for dette området var tydelig adskilt i forvaltningen, på samme måte som for andre GMO-søknader om utsetting. Dette for å sikre at risiko ble vurdert av fageksperter, og for å bidra til uavhengige vurderinger. Den generelle rutinen var at VKM vurderte søknaden om klinisk utprøving i to omganger. En første vurdering innen 45 dager, hvor søkers informasjon og dokumentasjon ble vurdert, og om denne var tilstrekkelig for å gjennomføre en risikovurdering. VKM utformet i denne perioden spørsmål om behov for tilleggsopplysninger fra søker. Endelig risikovurdering ble ferdigstilt når alle opplysninger fra søker var mottatt, innen 80 dager.
For søknader der søknadskjema for humane genmodifiserte celler kunne brukes, ba Miljødirektoratet VKM om å gjøre en forenklet risikovurdering, for å vurdere om søkers dokumentasjon var god nok til at kriteriet for standard miljørisikovurdering var oppfylt (se nærmere omtale i 12.2.3). Saksbehandlingstid på disse sakene var avtalt til maksimalt 45 dager, som tilsvarer en halvering av tiden i den generelle rutinen.
12.2.2.4 Ny grenseoppgang innesluttet bruk – utsetting
Tidligere godkjente Helsedirektoratet som hovedregel studier i sin helhet som innesluttet bruk. Miljødirektoratet ble involvert ved behov, og gjorde en skjønnsmessig vurdering av om studien ville innebære spredning/utsetting av GMO. Helsedirektoratet la i 2018 en strengere juridisk tolkning til grunn. Dette medførte at klinisk utprøving av GMO-legemidler kun kunne godkjennes i sin helhet som innesluttet bruk dersom hele studien ble gjennomført i fasiliteter godkjent for innesluttet bruk, ettersom genteknologilovens ordlyd fastsetter at all bruk som ikke er innesluttet, er utsetting. Forsøkspersonene kan med en slik tolkning ikke forlate fasiliteten godkjent for innesluttet bruk etter å ha mottatt GMO-legemidlet. Alle diskusjoner omkring dette omhandlet klinisk utprøving i mennesker.
Helsedirektoratet mente også at det med en slik strengere tolkning av loven ville være en byrde for søkere å måtte gjennomføre studien som helhet under innesluttet bruk-reglene, og at det ville være enklere om søker i stedet søkte godkjenning for GMO-legemidlet som utsetting:
GMM-ene har som regel lav helse- og miljørisiko. I mange tilfeller vil det være svært vanskelig, eller umulig å ivareta krav til innesluttet bruk for kliniske studier, bl.a. fordi innesluttet bruk av GMM skal foregå i godkjente laboratorier og anlegg. Det innebærer større praktisk ulempe å legge til rette for at kliniske studier vurderes som innesluttet bruk etter de krav som stilles i det regelverket, enn at det vurderes som utsetting (Helsedirektoratets avklaringsnotat om grenseoppgang innesluttet bruk og utsetting, 18. april 2019).
Det ble derfor etablert en forståelse om at søker for utprøving av GMO-legemidler til mennesker burde søke godkjenning for selve studien som utsetting hos miljømyndighetene. Det var ikke helt klart om utsettingsmyndigheten i lys av forskrift om innesluttet bruk av genmodifiserte mikroorganismer, kunne godkjenne genmodifiserte mikroorganismer til bruk i forskningsøyemed som utsetting. Innesluttet bruk-forskriften har som utgangspunkt at alt arbeid med genmodifiserte mikroorganismer skal skje i innesluttet bruk fasiliteter for å hindre unødvendig spredning av de genmodifiserte mikroorganismene og sikre et høyt beskyttelsesnivå. Andre land i EU (ti land) rapporterer også at de godkjenner prosedyrer på studiestedet som innesluttet bruk av genmodifiserte mikroorganismer i samsvar med direktiv 2009/41/EF om innesluttet bruk av genmodifiserte mikroorganismer.
Norske myndigheter landet derfor på en mellomløsning hvor Helsedirektoratet godkjenner innesluttet bruk i laboratorier, rom, anlegg og andre fasiliteter for følgende arbeid:
Mottak og lagring av GMO-legemiddel
Tilvirking av ferdig legemiddel om det ikke er ferdig til bruk (for eksempel fortynning av genmodifiserte virus vektorer i løsning i sprøyter, intravenøse væsker etc.)
Avhending og håndtering av rester av legemiddel og avfall som har vært i kontakt med GMO-en under disse prosedyrene
Å gi GMO-legemiddelet til pasient (intravenøst etc., dette kalles administrasjon)
Den kliniske utprøvingen godkjent som utsetting gjaldt dermed fra GMO-en ble gitt til forsøkspersonene og disse beveget seg ut fra de innesluttede fasilitetene, det vil si fra godkjente rom på sykehuset, til ut i korridor på sykehuset. Dersom Miljødirektoratet under risikovurderingen skulle identifisert en risiko ved GMO-en som ikke gjorde det forsvarlig å godkjenne studien som utsetting, skulle Helsedirektoratet og søker vurdere muligheten for om deler av studien kunne bli gjennomført som innesluttet bruk. Med forsvarlig i denne sammenheng menes det miljørisiko – dersom en GMO ville kunne bli vurdert å ha for stor risiko for spredning for eksempel ville det vurderes å ikke være forsvarlig å godkjenne hele studien som utsetting, men at deler av utprøvingen skulle skje under innesluttet bruk-reglene.
Helsedirektoratet har vært av den oppfatning at krav til innesluttet bruk etter genteknologiloven kan være ansett for strenge, og ikke formålstjenlige for klinisk utprøving av visse typer GMO-legemidler. Dette gjelder for eksempel humane genmodifiserte celler, hvor det kan dokumenteres at GMOen har en neglisjerbar miljørisiko. For slike studier vil også forskrift om håndtering av humane celler og vev komme til anvendelse10, og forskriften stiller krav til prosedyrer for uttak, prosessering, oppbevaring, distribusjon, mottak, emballering, merking og sporbarhet. I september 2021 var det dialog mellom Helsedirektoratet og Miljødirektoratet om en slik type studie, og det ble avklart at studien i sin helhet kunne godkjennes som utsetting. Det ble ikke stilt krav til søker etter regelverk om innesluttet bruk i genteknologiloven.
12.2.2.5 Felles postkasse
Etatene mente at det viktigste grepet for å sikre at søker fikk de godkjenninger de behøvde før planlagt oppstart av studien, var at søker søkte samtidig til alle etater.
Legemiddelverket og Helsedirektoratet tok i den forbindelse i 2018 initiativ til en felles postkasse (gmo@legemiddelverket.no) for søknader om kliniske utprøvinger med GMO-legemidler, som Miljødirektoratet ga sin tilslutning til i februar 2019. Hensikten var å forenkle søknadsprosessen for søker ved at alle søknader om godkjenning kunne sendes ett sted til alle etatene. Legemiddelverket driftet den felles postkassen og hadde førstekontakt med søker, og informerte søker og de andre etatene om søknader om klinisk utprøving av GMO-legemidler. Legemiddelverket skulle ved mottak av søknader etter genteknologiloven sende mottatte søknader til Miljødirektoratet og Helsedirektoratet.
I perioden 2019 – juli 2021 mottok Legemiddelverket en søknad til den felles postkassen (gmo@legemiddelverket.no). Søknaden etter legemiddelregelverket ble sent inn i februar 2019 på ordinær måte, mens søknaden etter genteknologiloven ble sendt til gmo@legemiddelverket.no i august 2019, og samtidig i kopi til Miljødirektoratet.
I juli 2021 mottok Legemiddelverket imidlertid en søknad om godkjenning både av studien etter legemiddelregelverket, om godkjenning av GMO-en etter genteknologiloven. Søker brukte ikke gmo@legemiddelverket.no, men Legemiddelverkets ordinære postkasse post@legemiddelverket.no. Søknaden om utsetting ble videreformidlet av Legemiddelverket til Miljødirektoratet i august 2021. Miljødirektoratets vurdering og endelige vedtak ble fattet før studien ble godkjent av Legemiddelverket og REK.
Den felles postkassen hadde frem til 2021 dermed ikke ført til samtidig innsendelse til alle etater, og koordineringen av søknadene fungerte ikke optimalt og etter hensikten. Etatene ser at en felles elektronisk søknadsløsning hadde vært bedre enn e-post. Med tanke på mengden søknader til nå har dette ikke blitt prioritert.
I forbindelse med overgang til sentral godkjenning av kliniske studier i EU/EØS etter ny forordning EU nr. 536/2014 om klinisk utprøving av legemidler til mennesker, må søkere fra og med 31. januar 2023 sende inn søknad etter legemiddelregelverk via en felles EU-portal (CTIS). Legemiddelverket, som etter 15. november 2021 er vedtaksmyndighet også etter genteknologiloven, vil fortsatt be søkere sende inn søknad etter genteknologiloven til gmo@legemiddelverket.no.
12.2.2.6 Informasjon og veiledning til søker
I tillegg til å legge til rette for en samtidig innsendelse av søknader under alle regelverk, arbeidet etatene med felles informasjon til søker som skulle legges ut på Legemiddelverket sine sider. Denne informasjonen skulle opplyse om krav til godkjenning av kliniske studier med GMO-legemidler, og også henvise søker til søkeprosedyrer via den felles postkassen. Arbeidet ble ledet av Legemiddelverket, men ble ikke ferdigstilt grunnet prioritering/kapasitet hos Legemiddelverket. Etatene oppdaterte derfor i stedet informasjon på sine respektive hjemmesider. Miljødirektoratet la ut informasjon og veiledning om hvordan søke om godkjenning til utsetting av GMO i kliniske studier på sine hjemmesider i juni 2019, og henviste her til den felles postkassen. Helsedirektoratet la i løpet av 2020 ut informasjon om GMO-legemidler på sine nettsider. Legemiddelverket la ut informasjon om godkjenning av GMO-legemidler og om felles postkasse i januar 2021.
12.2.2.7 Spesifikt om humane genmodifiserte celler
Vurderingen av humane genmodifiserte celler og genteknologilovens virkeområde har vært et diskutert, men ikke avklart spørsmål siden de første søknadene ble mottatt. Humane celler i kultur er ansett som organisme etter genteknologiloven, jamfør Ot.prp. nr. 8 (1992–93) om genteknologiloven s. 69. Transplantasjon av genmodifiserte, menneskelige organer omfattes ikke av loven. Genteknologi på mennesker er regulert under bioteknologiloven.11
Humane celler kan som hovedregel ikke overleve utenfor kroppen, med mindre de dyrkes i en spesialkultur in vitro. Hvorvidt de humane genmodifiserte cellene omfattes av genteknologiloven etter at de er satt tilbake i pasienten/studiedeltageren, er en gråsone i lovverket. I praksis har loven blitt forstått slik at cellene ikke lenger er omfattet etter innsettelse i studiedeltageren. Ved risikovurdering av de modifiserte cellene har fokuset dermed vært på de genmodifiserte virusvektorene som brukes til å modifisere cellene. En genmodifisert virusvektor vil alltid være omfattet av loven, uavhengig av om den befinner seg i celler i kultur eller i et menneske. Avhengig av egenskapene og framstillingen av den genmodifiserte virusvektoren, kan denne virusvektoren potensielt ha mulighet til å spre seg ut fra studiedeltageren og til miljøet, og eventuelt i ytterste konsekvens gi opphav til et nytt virus. Dette vurderes i en miljørisikovurdering.
Etter genmodifiseringen er det mulig at cellene kan inneholde (1) rester av replikasjonskompetent virusvektor12, og (2) resterende virusvektor-partikler.13 Et viktig vurderingspunkt har vært om søker kan dokumentere fravær av slike partikler.
Det var flere viktige avklaringer for genmodifiserte celler som ble gjort i løpet av 2018–2020 i EU.
I GMO-interplay arbeidet i EU ble det blant annet bekreftet at GMO-definisjonen etter utsettingsdirektivet i EU var lik den i Norge, det vil si at modifiserte celler i kultur er definert som GMO. Det ble utviklet et felles avklaringsdokument og felles søknadsskjema for modifiserte humane celler14, som ble diskutert mellom GMO-myndighetene Miljødirektoratet og Helsedirektoratet, samt Legemiddelverket. Myndighetene var enige i at søknadsskjemaet dekket kravene i genteknologiloven om godkjenning av kliniske studier med humane modifiserte celler som utsetting, og anbefalte at Norge ga sin tilslutning. For disse bestemte typene humane genmodifiserte celler var det faglig enighet om at disse kunne vurderes å ha neglisjerbar miljørisiko.
I 2020 ble det avklart at søkere skulle dokumentere et neglisjerbart nivå av resterende virusvektor-partikler i de genmodifiserte cellene, fremfor å skulle dokumentere fravær. Det var enighet om at det kan være utfordrende å bekrefte fullstendig fravær av virusvektorer, men at det er mulig å dokumentere et neglisjerbart nivå. I den forbindelse ble søknadskjemaet revidert.
Kymriah (genmodifiserte T-celler for CAR-T-behandling) ble søkt godkjent for markedsføring etter legemiddelloven, og det ble i den forbindelse utført miljørisikovurdering i 2018 i tråd med utsettingsdirektivets krav etter standard prosedyre for søknad om markedsføring av GMO-legemidler (se omtale av prosedyre i 12.4.1 og 6.3.1.3).
I lys av disse avklaringene var det norsk enighet om at det enkleste for søker var at kliniske utprøvinger med modifiserte humane celler kunne søkes godkjent ved hjelp av EU-skjema, og godkjennes som utsetting av GMO. Som nevnt i 12.2.2.4 ble en klinisk utprøving med humane genmodifiserte celler i september 2021 godkjent som utsetting i sin helhet, etter avklaring mellom Helsedirektoratet og Miljødirektoratet.
12.2.2.8 Om Miljødirektoratets saksbehandling som vedtaksmyndighet frem til november 2021
Saksbehandlingstiden hos Miljødirektoratet for de fem søknadene om klinisk utprøving av GMO-legemidler til mennesker som er godkjent som utsetting av GMO etter genteknologiloven, var mellom 80 og 120 virkedager for de fire første (lengste saksbehandlingstid for den første søknaden). Siste søknad mottatt i august 2021 hadde en saksbehandlingstid på en drøy måned. Hovedsakelig gikk saksbehandlingstiden med til miljørisikovurderingen gjennomført hos VKM. Saksbehandlingstiden for utsetting etter genteknologiloven har vært nokså lik saksbehandlingstiden etter legemiddelregelverket som både REK og Legemiddelverket følger. Ifølge saksbehandlingsreglene i rådsdirektiv nr. 2001/18/EF (utsettingsdirektivet) artikkel 6, og forskrift om konsekvensutredning etter genteknologiloven § 8 starter saksbehandlingstiden ved den datoen søknad mottas. Imidlertid stoppes klokken den tiden myndighetene venter på manglende/tilleggsinformasjon fra søker som myndigheten har bedt søker om å fremskaffe. Full saksbehandlingstid tilgjengelig etter forskrift om konsekvensutredning etter genteknologiloven og utsettingsdirektivet er 90 virkedager + 30 virkedager for høring (120 virkedager totalt).
Så lenge Miljødirektoratet var vedtaksmyndighet, ga direktoratet tillatelse til klinisk utprøving av GMO-legemiddel til utsetting etter genteknologiloven for alle mottatte søknader. Tillatelsene ble gitt med bakgrunn i informasjonen fra søker, VKMs helse- og miljørisikovurdering om neglisjerbar/lav risiko ved utsetting i de konkrete saker, og eventuelt innkomne høringsinnspill. Kun vilkår om rapportering, jamfør forskrift om konsekvensutredning etter genteknologiloven § 18 første ledd, ble gitt i tillatelsene, ettersom det ble vurdert at andre vilkår ikke var påkrevd. I tråd med gjeldende praksis ble det også gitt tillatelser av Helsedirektoratet etter regelverk om innesluttet bruk.
Frem til 8. juni 2020, da Stortinget vedtok endringer i bioteknologiloven, vurderte Bioteknologirådet søknader om GMO-legemidler til bruk som genterapi etter bioteknologiloven. Bioteknologirådet uttaler seg også etter eget initiativ om samfunnsnytte, bærekraft og etikk etter genteknologiloven i forbindelse med høringer.
Legemiddelverket har fra og med 15. november 2021 hatt vedtaksmyndighet for søknader om klinisk utprøving av GMO legemidler til mennesker og dyr, etter genteknologiloven § 10 første ledd. Myndighetsoverføringen følger av kgl.res. 3. september 2021. Det kommer frem av side 2 at ordningen skal gjelde frem til forslagene fra GMO-utvalget er lagt frem og er nærmere vurdert. Det fremgår også at ved godkjenning skal det innhentes uttalelse fra Miljødirektoratet som grunnlag for beslutningen, og i miljørisikovurderingen skal det legges avgjørende vekt på uttalelsene fra miljømyndighetene.
12.2.3 Søknadsskjema og veiledning utarbeidet i EU – GMO-interplay status i 2022
Under GMO-interplay arbeidet har det blitt utviklet veiledninger og søknadsskjema. Disse er frivillige å ta i bruk i hvert enkelt land, og søknadskjemaene kan brukes etter regelverk om både innesluttet bruk og utsetting. Det angis hvilke land søknadsskjemaene kan benyttes i. Veiledningene og skjemaene oppfattes som forenklende både for søker og myndigheter.
Søknadsskjema og veiledningsdokument for genmodifiserte humane celler har i flere runder blitt revidert til å inkludere flere produktgrupper, og i sammenheng med dette har landene blitt bedt om å vurdere tilslutning på nytt. Siste runde med revisjon var i 2021, og det har ikke blitt avholdt møter i GMO-interplay pharmagruppen etter juni 2021. Andre dokumenter har blitt oppdatert ved at nye land har tilsluttet seg. Slike oppdateringer kan sees i oversikten på dokumentenes første side.
Med avklaringene gjort mellom Helsedirektoratet og Miljødirektoratet i 2018 angående utsetting, har søknadsskjemaer først og fremst vært tenkt brukt til å søke godkjenning som utsetting. Naturlig nok, ettersom dette er særnorske krav, inneholder ikke søknadskjemaene spørsmål om vurdering av bærekraft, samfunnsnytte og etikk. I påvente av eventuell endring av genteknologiloven angående vurderingen av bærekraft, samfunnsnytte og etikk, har det siden 2020 heller ikke blitt bedt om en vurdering fra søker for disse kriteriene.
De aller fleste medlemslandene har gitt sin tilslutning til alle GMO-interplay dokumentene. Før september 2021 hadde Norge formelt tilsluttet seg, eller godkjent for bruk, fem av ni dokumenter for klinisk utprøving med GMO-legemidler til mennesker. I september 2021 ble søknadskjemaet og veiledningsdokumentet for humane genmodifiserte celler revidert til også å inkludere genom-redigerte humane celler. I sammenheng med at Norge ga tilslutning til denne revisjonen, meldte Miljødirektoratet også til EU-kommisjonen at Norge tilsluttet seg alle søknadskjema for GMO-legemidler og tilhørende dokumenter. En oversikt over alle søknadskjema og veiledningsdokumenter kan finnes på EU-kommisjonens nettsider15.
Miljødirektoratet ga VKM i oppdrag å vurdere en del av søknadskjemaene og veiledningsdokumentene, i perioden Miljødirektoratet var vedtaksmyndighet. VKM konkluderte med at alle søknadskjemaene etterspør tilstrekkelig informasjon fra søker til at miljørisiko av GMO-legemiddelet kan vurderes16.
To av søknadskjemaene kommer med en tilhørende ferdig utfylt miljørisikovurdering. Dette er skjemaene til bruk for bestemte typer humane genmodifiserte celler, og for adeno-assosierte virusvektorer (AAV-vektorer). Det er faglig enighet om at disse produktgruppene har neglisjerbar miljørisiko dersom en del kriterier er oppfylt17. Søker må altså kunne dokumentere at kriteriene er oppfylt for å kunne benytte søknadskjemaet og vise til en ferdig utfylt miljørisikovurdering. Med en ferdig utfylt miljørisikovurdering, vil risikovurderingen av GMOen benyttet i slike kliniske studier i hovedsak bestå av gjennomgang av søkers dokumentasjon, og således tilsvare en forenklet miljørisikovurdering som reduserer saksbehandlingstiden.
I juni 2021 sendte EU-kommisjonen en forespørsel om å gi tilbakemelding om landenes erfaring med klinisk utprøving av GMO-legemidler til mennesker. EU-kommisjonen opplyste at henvendelsen kom i tilknytning til arbeidet med legemiddelstrategien og mulige endringer i regelverk for klinisk utprøving. Miljømyndighetene i Norge valgte å svare relativt kort på denne henvendelsen, basert på at Miljødirektoratet har behandlet få søknader etter genteknologiloven.
I denne sammenheng opplyste Nederland at de jobbet med å utvikle flere søknadskjema for spesifikke produktgrupper, ettersom de fikk mer erfaring med kliniske studier med flere typer GMO-legemidler. Disse søknadskjemaene utarbeides av risikovurderingsorganet COGEM. COGEM har vurdert at man ikke kan sette kriterier på forhånd til hvilke GMO-legemidler som kan vurderes til å ha en neglisjerbar miljørisiko, men at dette må være en læringsprosess der man kan utvikle søknadskjema med slike ferdigutfylte, standardiserte miljørisikovurderinger etter hvert som man opparbeider seg erfaring med forskjellige typer GMO-legemidler18.
12.3 Sammenligning av norsk praksis for klinisk utprøving av GMO-legemidler til mennesker med øvrig praksis i EØS-området
12.3.1 Generelt om praksis i EU/EØS
Praksis for GMO-godkjenning av utprøving av GMO-legemidler er heterogen i EØS-området, hvor landene godkjenner klinisk utprøving av legemidler til mennesker enten som utsetting, innesluttet bruk, eller begge. Det er også flere myndigheter involvert i mange EØS-land. Dette medfører et uoversiktlig regulatorisk bilde for søkere som ønsker å søke godkjenning for studier som skal gjennomføres i flere land i EU og utenfor EU19.
Med oppstarten av interplay-gruppene i 2017 i EU/EØS ble landene oppfordret til å gi informasjon om sine nasjonale prosedyrer for godkjenning av GMO til klinisk utprøving. I 2021 ba EU-kommisjonen landene om å oppdatere denne informasjonen. Basert på disse opplysningene, som er publisert på EU-kommisjonens hjemmesider20, har utvalget sett på hvilke ulike modeller man har for praktisering i hele EØS-området. De ulike GMO-godkjenningsmodellene i EØS-området er illustrert i figuren under.
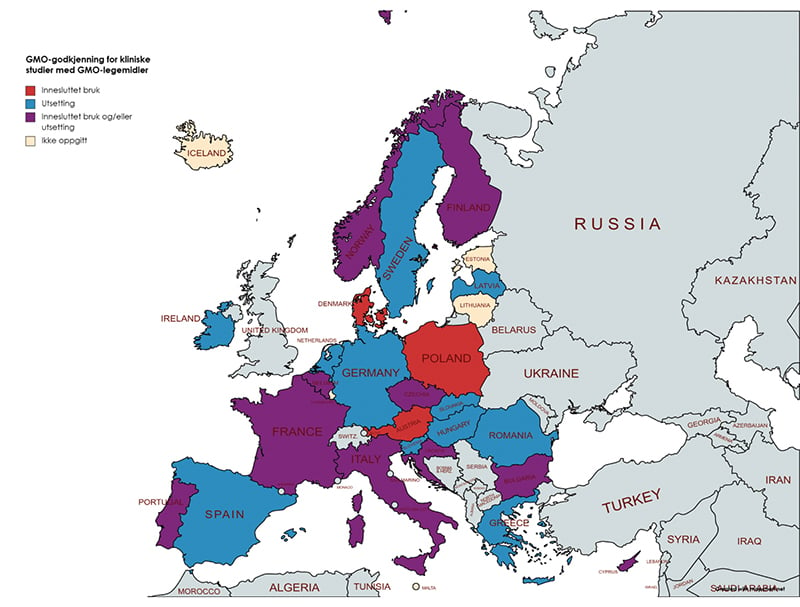
Figur 12.2 Oversikt over praksis etter GMO-regelverket for godkjenning av GMO i legemidler til klinisk utprøving i mennesker i EØS-området
Informasjon viser at noen EU/EØS-land godkjenner GMO-legemidler i klinisk utprøving som utsetting, noen som innesluttet bruk og andre etter begge regelverkene (norsk modell). Kun tre land opplyser å godkjenne klinisk utprøving av GMO-legemidler som innesluttet bruk. For fem land er det ikke gitt opplysninger om hvilket GMO-regelverk GMO-legemidler til klinisk utprøving godkjennes etter.
12.3.2 Godkjenning kun som utsetting av GMO
En knapp majoritet (11 land) oppgir at kliniske studier godkjennes som utsetting i samsvar med kravene i utsettingsdirektivet. Samtlige land, unntatt Hellas, oppgir at det gjennomføres høring av GMO-søknadene. Form og innhold i høringene varierer imidlertid, jamfør kapittel 12.3.5.
Landene har ikke omtalt om de medregner mottak, lagring og tilvirking av GMO på studiestedet i en slik utsettingsgodkjenning, eller om dette ikke regnes som en del av selve den kliniske studien.
12.3.3 Innesluttet bruk og/eller utsetting
Ti land, inkludert Norge, oppgir å godkjenne kliniske studier som innesluttet bruk som hovedregel (arbeid, håndtering, lagring og å gi medikamentet til pasient), men også som utsetting avhengig av saken.
Det er noen nyanseforskjeller i praksis mellom landene som oppgir å ha denne modellen.
I Frankrike og Italia vurderer innesluttet bruk myndigheten om søknaden medfører utsetting av GMO i miljøet. Søknader om GMO-legemiddelutprøving sendes da først og fremst til innesluttet bruk myndigheten for godkjenning av produksjon, mottak, håndtering, avhending og administrasjon til pasienten. Dersom disse finner at det er sannsynlig at GMO-en vil avgis til og spres i omgivelsene (GMO-en er formeringsdyktig) gjennom studien, skal en søknad også sendes miljømyndighetene som vurderer miljørisiko/miljørisikohåndteringstiltak og er godkjenningsmyndighet for utsetting.
I Finland er GMO-myndighetene koordinert gjennom Board of Gene Technology, som vurderer og er godkjenningsmyndighet både for innesluttet bruk og utsetting av GMO-legemidler. Det vurderes fra sak til sak om søknaden skal behandles kun etter innesluttet bruk reglene, eller også som utsetting, basert blant annet på egenskapene til GMO-en, for eksempel om den består av replikasjonskompetent (formeringsdyktig) virus.
I Norge kan man sammenligne praksis før 2018 med en slik modell hvor kliniske studier med GMO-legemidler før 2018 i praksis kun ble godkjent som innesluttet bruk, men hvor miljømyndighetene ble forespurt i saker hvor Helsedirektoratet mente det kunne være en mulighet for utsetting av GMO gjennom oppsettet av studien og egenskapen til GMO-en. Miljødirektoratet gjorde da en forhåndsvurdering av om studien ville medføre utsetting eller ikke, og om søknad om utsetting ville være nødvendig.
Land som gir opplysninger om hvordan de vurderer om godkjenning til utsetting av GMO er nødvendig ved legemiddelutprøvingen, oppgir at dette er nødvendig dersom GMO-en:
1. kan replikere seg (formeringsdyktig)
2. avgis (shedding fra testpersonen)
3. spres i miljøet (tas opp av andre mennesker og/eller dyr, kan overleve i omgivelser inne/ute/på overflater/i avløpsvann, jord med mer)
Noen land som Italia oppgir at alle kriteriene må være oppfylt for at søknad om utsetting er nødvendig, mens noen angir bare et eller to som eksempler på kriterier som medfører at godkjenning som utsetting er et krav. Dersom en slik praksis skulle være aktuelle i Norge, bør det defineres og klargjøres for søker hvilke kriterier som skal ligge til grunn for godkjenning og hvilken dokumentasjon som skal sendes inn.
12.3.4 Godkjenning kun som innesluttet bruk
Tre land i EØS-området oppgir å godkjenne kliniske utprøvinger med GMO-legemidler kun etter reglene for innesluttet bruk; Danmark, Polen og Østerrike. I Danmark og Polen er det miljømyndighetene som er kompetent GMO-myndighet for innesluttet bruk, og som er ansvarlig for å godkjenne den innesluttede bruken. I Østerrike er det helsemyndighetene som er ansvarlig GMO-myndighet for innesluttet bruk (Federal Ministry of Social Affairs, Health, Nursing and Consumer Protection).
Kun Polen opplyser å gjennomføre høring av søknader om innesluttet bruk av GMO i legemiddelutprøving.
Formålet med innesluttet bruk regelverket er å forhindre at GMO slipper ut til omgivelsene og miljøet, for å sikre et høyt beskyttelsesnivå av mennesker, dyr og miljøet. En forutsetning for en slik modell vil dermed være at man legger til grunn at klinisk utprøving av GMO-legemidler uavhengig av type GMO og bruksområde, ikke resulterer i at GMO slipper ut fra pasienten via avgivelse fra kroppsvæsker.
12.3.5 En felles innsendelse av søknader til en instans, og en felles godkjenning
Seks av landene som oppgir å godkjenne GMO-legemidler kun som utsetting, oppgir også å ha én felles innsendelse av søknad til én myndighet (single submission procedure), til legemiddelmyndigheten. GMO-søknaden legges da ved søknaden om klinisk utprøving. Det fattes et felles vedtak som utstedes samtidig til søker for både den kliniske studien og GMO-søknaden av legemiddelmyndigheten. Dette gjelder Sverige, Tyskland, Litauen, Ungarn, Estland, og Hellas. I tillegg opplyser Belgia, at det ved godkjenning som utsetting, er legemiddelmyndigheten som har fått delegert vedtaksmyndigheten for GMO-søknader, men at vurderingene gjøres av et Biosikkerhetsorgan.
I de landene som har oppgitt mest informasjon om denne modellen virker det å være en intern prosedyre mellom etatene, som ikke involverer søker. Søker forholder seg derfor kun til en myndighet, den som godkjenner selve den kliniske studien. GMO-myndighetene vurderer eller gir råd om risiko av GMO-søknaden før et felles vedtak etter begge regelverk utstedes av legemiddelmyndigheten.
Med endringen i overføringen av vedtaksmyndighet fra Miljødirektoratet til Legemiddelverket, kan ordningen i Norge sies å nærme seg den presentert over, med unntak av at utprøvinger ikke kun godkjennes som utsetting, og at Helsedirektoratet fortsatt er godkjenningsmyndighet for innesluttet bruk.
I to av disse landene, Sverige og Tyskland, som har denne modellen, har man et annet system for høring av søknaden. I Tyskland, gjøres kun informasjon om søknaden offentlig tilgjengelig på hjemmesiden til legemiddelgodkjenneren (Paul Ehrlich Institute) etter vedtak om godkjenning er fattet. I Sverige, legges informasjon om søknaden også kun ut på hjemmesiden til Läkemedelsverket (uten kunngjøring eller frist for innspill som ved høring). Sverige regner legemiddelmyndighetens konsultasjon av GMO-myndighetene om GMO-søknaden som å dekke kravet til høring av GMO-søknadene. I Norge har vurderingen vært at søknader om utsetting etter genteknologiloven må på høring, og at man ikke kan anse at Miljødirektoratets uttalelse om miljørisikovurdering dekker utsettingsdirektivets krav til høring.
12.3.6 Saksbehandlingstid og høring
Det er ikke særskilt opplyst om saksbehandlingstid i nasjonale skjema for EU-landene. For søknader etter utsettingsdirektivet er det 90 dager, pluss eventuelle 30 dager til høring, som er det de fleste land har oppgitt. Nederland delte i august 2021 detaljert informasjon om sin saksbehandling på området, og har utviklet prosedyrer på 56 kalenderdager for visse typer GMO-er med kjent neglisjerbar risiko. Norge har som tidligere nevnt (kapittel 6.3.2) rutiner på at saksbehandling etter nasjonalt regelverk og ny forordning (EU) nr. 536/2014 for klinisk utprøving til mennesker skal samkjøres, og det skal fattes vedtak på samme tid så langt det er mulig. Det vil si tidligst etter 55 dager. Godkjenning av studier etter legemiddelregelverket har gitte frister, men disse er dynamiske avhengig av blant annet når søker svarer.
Flertallet av EU-land opplyser at de gjennomfører høring ved søknader om utsetting av GMO. De fleste oppgir at dette ikke skal forlenge saksbehandlingstiden etter GMO-regelverk med mer enn 30 dager. Intensjonen med nye rutiner i Norge er at høringen ikke skal forlenge saksbehandlingen etter hverken legemiddelregelverk eller genteknologiloven. Norsk praksis om høring er dermed sammenlignbar med de fleste andre EU-land. Ett land, Finland, opplyser å gjennomføre 60 dagers høring.
12.4 Om markedsføringstillatelser (omsetting av GMO-legemidler)
12.4.1 Markedsføringstillatelse (omsetning) for legemidler til mennesker
Legemiddelloven og legemiddelforskriften implementerer de mest relevante EU-rettsaktene, direktiv 2001/83/EF og forordningene (EF) nr. 726/2004 og (EF) nr. 1394/2007 for regulering av markedsføringstillatelser. Sistnevnte er et tillegg til direktiv 2001/83/EF og omhandler legemidler til avansert terapi. Et legemiddel blir bare godkjent for salg dersom legemiddelet har en nytte som overstiger risikoen ved bruk. Vurdering av risiko/nytte-forholdet til et legemiddel er basert på dokumentasjon som produsentene må sende inn når de søker om markedsføringstillatelse. I søknaden må produsenten dokumentere legemidlets farmasøytiske kvalitet, sikkerhet og medisinske effekt. Godkjenningsprosessen skjer i de fleste tilfeller i samarbeid med andre europeiske legemiddelmyndigheter, enten som en såkalt sentral prosedyre, eller som en desentralisert prosedyre.
Alle nye legemidler, inkludert GMO-legemidler, skal søkes godkjent i sentral prosedyre, slik at markedsføringstillatelsen (vilkårene for omsetting) blir lik i alle EØS-land. I forbindelse med vurdering av risiko og nytten til legemidlet når det søkes om markedsføringstillatelser, vurderes også miljørisikoen for legemidlet. For legemidler som inneholder GMO skal miljørisikovurderingen utføres i henhold til kravene for miljørisikovurdering av GMO gitt i utsettingsdirektivet, og det er satt opp en GMO-prosedyre for legemidler som skal ivareta denne, beskrevet i kapittel 6.3.1.3.3.
I 2008 tok Klima- og miljødepartementet og andre berørte departementer stilling til forholdet mellom genteknologiloven og legemiddelloven når det gjaldt GMO-legemidler til mennesker og dyr. Det ble da bestemt at søknad om markedsføringstillatelse av GMO-legemidler ikke lenger skulle godkjennes etter genteknologiloven, men etter legemiddelregelverket. Dette er i tråd med unntaksadgangen i utsettingsdirektivet art. 12. Norsk praksis for markedsføringstillatelse er dermed i overensstemmelse med praksis i EØS. Godkjenningen etter legemiddelregelverket, der Legemiddelverket deltar i godkjenningsprosedyren, baseres på en vurdering av om legemiddelet oppfyller kravene til kvalitet, sikkerhet og effekt og en miljørisikovurdering (sistnevnte på grunnlag av kravene i utsettingsdirektivet). Det gjøres også en miljørisikovurdering etter legemiddelregelverket. Praksis siden 2008 for behandlingen av søknader om markedsføring av GMO-legemidler har dermed vært at Legemiddelverket som myndighet etter legemiddelregelverket, har ansvar for vurdering av legemidlet gjennom EØS-prosedyren for markedsføringstillatelse. Miljødirektoratet, etter instruks fra Klima- og miljødepartementet, følger prosedyren for høring av GMO-myndighetene angående miljørisikovurderingen av GMO-legemidlet til markedsføring. Etter denne prosedyren sender Det europeiske legemiddelbyrået (EMA) miljødelen av søknader på høring til myndighetene under utsettingsdirektivet. Miljødirektoratet gir innspill om eventuell miljørisiko og miljørisikohåndteringstiltak for GMO-legemidlet direkte til EMA, med kopi til Legemiddelverket. Innspillene fra Miljødirektoratet bygger på risikovurderinger fra Vitenskapskomiteen for mat og miljø (VKM).
12.4.2 Markedsføringstillatelse (omsetting) for legemidler til dyr
Fra 28. januar 2022 kom den nye forordningen om legemidler til dyr (forordning (EU) nr. 2019/6) til anvendelse i EU/EØS. Endringen medførte en overgang fra direktiv til forordning. Dette betyr samme regelverkstekst i alle land, noe som reduserer nasjonale forskjeller. Forordningen representerer hovedrettsakten på området legemidler til dyr, og er implementert som lov i Norge (i legemiddelloven). Flere sekundærrettsakter fikk også anvendelse fra samme tidspunkt. Disse gir mer detaljerte bestemmelser på enkelte punkter. Ytterligere sekundærrettsakter vil komme etter hvert. En del legemidler til fisk søkes imidlertid godkjent nasjonalt i Norge. Alle nye legemidler som er fremstilt ved hjelp av bioteknologi, inkludert GMO-legemidler skal søkes godkjent i sentral prosedyre, slik at markedsføringstillatelsen (vilkårene for omsetting) blir lik i alle EØS-land.
Legemiddelmyndighetene vurderer dokumentasjon av kvalitet, sikkerhet og effekt som søker legger ved søknaden. Sikkerheten til personer som skal håndtere legemidlene, vurderes også. For legemidler til matproduserende dyr er sikkerheten til konsumenter som spiser matvarer fra behandlede dyr, et viktig aspekt.
Alle som produserer, importerer og omsetter legemidler til dyr, må også ha godkjenning fra Legemiddelverket. Legemiddelverket vurderer også søknader om forskrivning av legemidler som krever spesiell tillatelse til rekvirering.
Mattilsynet er ansvarlig myndighet for bruk av legemidlene og fører tilsyn med at dyrehelsepersonell bruker legemidler forsvarlig.
I forbindelse med vurdering av risikoen og nytten til legemidlet, vurderes også miljørisikoen for legemidlet. For legemidler som inneholder GMO skal miljørisikovurderingen utføres i henhold til kravene for miljørisikovurdering av GMO gitt i utsettingsdirektivet, og det er satt opp en GMO-prosedyre for legemidler som skal ivareta denne, beskrevet i kapittel 6.3.1.3.3.
12.5 Klinisk utprøving av GMO-legemidler til dyr
Det har til nå ikke vært noen kliniske utprøvinger med GMO-legemidler til dyr i Norge. Fra Norges side har det blitt prioritert å følge det europeiske GMO-interplay arbeidet for klinisk utprøving til mennesker. Det er fra norske myndigheters side ikke kjent om, eller i hvilken grad, EU/EØS-landene har ulik praksis når det gjelder godkjenning av klinisk utprøving av legemidler til dyr vs. mennesker.
Legemiddelregelverket, både nasjonalt og i EU/EØS, er delt opp slik at det er forskjellig regelverk og dermed forskjellige krav til klinisk utprøving av legemidler i mennesker og i dyr. Dette kan man anse som hensiktsmessig da legemiddelregelverket har som formål å ivareta testsubjektenes hensyn. Det vil være store forskjeller i hvordan man skal sette opp en klinisk utprøving og ivareta hensynet til testsubjektene, avhengig av om disse er mennesker eller dyr, og hva formålet med bruken av legemidlene er. I regelverket skilles det også mellom klinisk utprøving av legemidler til dyr (legemiddelregelverket), og prekliniske forsøk (forsøksdyrforskriften). Se nærmere om forsøksdyr-regelverket og forsøksdyrforskriften i 12.5.1 og 12.5.2.
Et slikt skille har man ikke i GMO-regelverket, man vurderer GMO-en i legemidlet, som gjerne er mikroorganismer. Fokuset er på GMM-en, og eventuelle effekter den har på miljøet rundt (som også omfatter andre mennesker enn testsubjektet). Kravene til vurdering av genmodifiserte mikroorganismer er i utgangspunktet likt, uavhengig av om mottaker er mennesker eller dyr. Det vil naturligvis være forskjell i vurderinger rundt innesluttet bruk, ettersom mennesker alltid vil kunne forlate innesluttet-fasiliteten i en klinisk utprøving, mens visse utprøvinger i dyr vil kunne gjøres innesluttet i sin helhet. Oppsettet og hvor den kliniske utprøvingen foregår, er også som regel ulikt. Klinisk utprøving av GMO-legemidler til mennesker skjer i all hovedsak på et sykehus, med oppfølging utenfor innesluttet-fasiliteten, mens klinisk utprøving av GMO-legemidler til dyr kan skje hos veterinæren eller i normale dyrehold. Det kan også variere hvem som håndterer GMO-legemidlet, om det er en veterinær eller bonde, og hvordan GMO-legemidlet administreres, som spray i munn/nese, eller som injeksjon. Disse momentene skriver EMA om i sin veiledning for miljørisikovurdering av veterinære GMO-legemidler21. Videre opplyses det i veiledningen om at GMO-ens evne til å overleve over lengre perioder i miljøet (for eksempel i avføring i fjøset eller på beitemark) kan utgjøre en fare under noen omstendigheter, for eksempel hvis det kan bety at det er større sannsynlighet for kontakt med enkeltpersoner/andre dyr. Dette avhenger av hvilken dyreart som benyttes i den kliniske utprøvingen (det finnes på markedet i EU/EØS GMO-vaksiner til fjørfe som holdes innomhus, men ikke til frittgående beitedyr).
Hva som skal vurderes i en risikovurdering for GMO-aspekter varierer fra sak til sak, og mellom klinisk utprøving til dyr sammenlignet med til mennesker. Dersom gjødselhåndteringen inaktiverer GMO-vaksinerester, kan det vurderes at den reelle eksponeringen til miljøet sannsynligvis i praksis ikke er så forskjellig mellom klinisk utprøving til mennesker eller dyr. Denne vurderingen gjelder imidlertid ikke for akvatiske dyr, for eksempel fisk.
12.5.1 Hva omfattes av klinisk utprøving av legemidler til dyr?
Klinisk utprøving til dyr omfatter ikke grunnforskning eller pre-kliniske forsøk etter legemiddelregelverk. Klinisk utprøving av veterinære legemidler på dyr er etter legemiddelregelverk definert som stadiet etter det prekliniske forsøksstadiet. I motsetning til klinisk utprøving omfattes pre-kliniske forsøk av forsøksdyrregelverket (se 12.5.2). Etter genteknologiloven har man ikke et slikt skille mellom forskning/forsøk og klinisk utprøving, her reguleres begge bruksområder etter samme lov.
Ved klinisk utprøving av vaksiner til dyr må legemiddelprodusenten avklare med Mattilsynet om det må innhentes brukstillatelse fra Mattilsynet før vaksinen kan tilføres norske dyr.
For klinisk utprøving av vaksiner til fisk er det egne retningslinjer22. Definisjonen av klinisk utprøvning omfatter ikke karforsøk, og normalt ikke minimerdforsøk, utført på forskningsstasjoner med tillatelse til å drive forsøk med fisk, med mindre forsøksfisken i slike undersøkelser senere skal benyttes til matproduksjon. Forsøk der utsett av vaksinert fisk i minimerd ved forskningsstasjoner kan tenkes å medføre miljømessige konsekvenser (for eksempel levende vaksine uten markedsføringstillatelse i Norge), omfattes likevel av definisjonen. I tvilstilfeller avgjør Statens legemiddelverk om et forsøk skal regnes som klinisk utprøvning eller laboratorieforsøk.
12.5.2 Nærmere om tilgrensende regelverk – forsøksdyrforskriften
Før en virksomhet kan søke om godkjenning for klinisk utprøving av et legemiddel, gjøres en rekke grunnforskningsforsøk og pre-kliniske forsøk. Dataene innsamlet i forbindelse med disse forsøkene hvor det brukes forsøksdyr, er basis for senere søknad om klinisk utprøving etter legemiddellovgivningen, og i tillegg etter genteknologiloven dersom legemidlene består av GMO.
Når det gjelder forsøksdyrsforskriften, gjelder denne for pre-kliniske forsøk både for legemidler til mennesker og dyr, herunder GMO-legemidler, og for andre forsøk. Imidlertid kommer forskriften ikke til anvendelse ved godkjenning av kliniske utprøvinger med legemidler til dyr.
Hvis den kliniske utprøvingen har vært på matproduserende dyr, kan dyrene etter ferdig utprøving og etter en eventuell tilbakeholdelsestid for legemiddelet, som oftest gå inn i matkjeden. Dette til forskjell fra forsøksdyr brukt i grunnforskning og pre-kliniske forsøk, hvor dyrene oftest avlives og skrottene destrueres ved godkjente destruksjonsanlegg etter at forsøkene er ferdige.
Forsøksdyrforskriften og instruks om Mattilsynets forvaltning av forsøksdyrforskriften23, gjennomfører til sammen direktiv 2010/63/EU om bruk av dyr til vitenskapelige formål. Mattilsynet skal godkjenne alle virksomheter som driver med forsøksdyr i Norge, og behandler de enkelte søknadene om forsøk og bruken av forsøksdyr.
Det følger av forsøksdyrforskriften § 5 at oppdrettere, formidlere og brukere, og lokalene de bruker, skal være godkjent av Mattilsynet. Videre følger det av § 6 at Dyr kan brukes i forsøk bare hvis Mattilsynet har godkjent forsøket. Det er ikke lov å benytte dyr i forsøk hvis man kan oppnå samme kunnskap uten bruk av dyr. Det skal alltid benyttes så få dyr som mulig, og forsøket skal alltid utføres slik at dyrevelferden er best mulig under forsøket.
Hver søknad om dyreforsøk blir vurdert av Mattilsynet ut fra en kost-/nyttevurdering der de 3R-ene Erstatning (Replacement), Reduksjon (Reduction) og Forbedring (Refinement) er helt sentrale, og alle søkere må greie ut om disse i søknadsskjemaet. Mattilsynet har oppnevnt eksterne eksperter som benyttes som rådgivere i spesielt vanskelige søknader. Regjeringen har dessuten opprettet en Nasjonal komité for beskyttelse av forsøksdyr (forsøksdyrkomitéen24) i tråd med EUs forsøksdirektiv. Denne komitéen skal være et rådgivende organ for forvaltningen og de enkelte forsøksdyrvirksomhetene.
Planer om dyreforsøk må kvalitetssikres på minst tre ulike nivåer: de skal være lovlige, de skal være av høy vitenskapelig kvalitet og de skal være etisk forsvarlige. Det siste er naturlig nok det vanskeligste, men vitenskapelige standarder er heller ikke alltid lett å enes om. For å veie opp verdien av et forsøk mot belastningen på dyret, må det foretas en nytte-kostnadsanalyse. Forsøkets samfunnsnytte samt etiske forhold rundt bruken av forsøksdyr, er viktige prinsipper som vurderes av Mattilsynet i forbindelse med godkjenning av søknad om bruk av forsøksdyr.
Den nasjonale forskningsetiske komité for naturvitenskap og teknologi (NENT) har utarbeidet etiske retningslinjer for bruk av dyr i forskning25, med formål om å gi etisk veiledning til forskere og andre som vurderer dyreforsøk.
Antallet søknader til Mattilsynet om bruk av dyr til forsøk har de siste årene ligget på rundt 600 per år. Mattilsynets rutine er at søker kontaktes og veiledes ved uklarheter eller mangler i søknaden, og svært få søknader blir godkjent uten denne dialogen og veiledningen. I 2022 var det ca. 80 virksomheter med egne forsøksdyravdelinger i Norge. I tillegg gjøres det forsøk med ville dyr i felt. Rundt 95 % av dyrene brukt i godkjente dyreforsøk i 2020 var fisk, hvor laks utgjorde den klart største gruppen.
For mer utfyllende informasjon om forsøksdyrforskriften, og Mattilsynets rolle i vurdering og godkjenning av bruk av forsøksdyr, henvises det til kapittel 6 om dagens regulering og forvaltning av genmodifiserte organismer og avlede produkter.
12.5.3 Om norsk erfaring med klinisk utprøving av GMO-legemidler til dyr
De GMO-legemidler som til nå er godkjent til dyr i EU/EØS er profylaktiske vaksiner til landdyr og fugler. En DNA-vaksine mot pankreassykdom hos laks er også godkjent. Ingen av disse har vært i klinisk utprøving i Norge, og Legemiddelverket har derfor ingen erfaring med å vurdere de aspektene som er spesielle for utprøving av slike legemidler.
Miljømyndighetene i EU involveres i søknader om markedsføringstillatelser for GMO-legemidler og får tilsendt relevante deler av dokumentasjonen. Miljørisikovurdering er et krav etter direktiv 2001/18/EF, og dette foregår som en høring i regi av det europeiske legemiddelbyrået EMA (se omtale i kapittel 6.3.1.3.3). Norske miljømyndigheter svarer på høringene og har derfor erfaring i vurdering av aspektene for GMO-vaksiner til dyr, herunder risiko for spredning til ikke-målorganismer.
12.5.4 Vurderes hensyn til bærekraft, samfunnsnytte og etikk etter tilgrensende regelverk?
Legemiddelverket vurderer utprøvingens risiko/nytte-forhold og forsvarlighet. Det ultimate formålet med en legemiddelutprøving er å fremskaffe data om legemidler, og dermed om legemidlet kan brukes i behandling av sykdom i fremtiden. Det vil ikke godkjennes utprøvinger som ikke vil ansees som samfunnsnyttige og etisk forsvarlige. Bærekraftig utvikling slik det forstås i genteknologiloven er ikke spesifisert i regelverkstekst og er ikke et vilkår.
Forsøksdyrforskriften §§ 8-11, § 24 og vedlegg E hjemler vurdering av samfunnsmessig nytteverdi og etikk for ethvert forsøk som innebærer bruk av dyr i grunnforskning og pre-kliniske forsøk. Bærekraftig utvikling er ikke omtalt i forskriften eller Mattilsynets instruks.
Relevant å nevne i denne sammenheng er at Norge og EU generelt har et meget restriktivt regelverk som begrenser godkjenning og bruk av veterinære legemidler, uavhengig av om de er GMO-baserte eller ikke. Dette medfører at flere typer veterinære legemidler ikke kan godkjennes eller brukes i Norge og EU/EØS til tross for omfattende bruk internasjonalt.26
12.6 Vurdering av praksis og mulige effektiviseringsgrep for klinisk utprøving av GMO-legemidler til mennesker og dyr
Legemiddelfirma som skal gjennomføre kliniske utprøvinger i et land legger blant annet vekt på kriterier som tilgang til pasienter, tilgang til studiesteder og rask saksbehandlingstid/regulatoriske forhold. I handlingsplaner i Norge og EU om kliniske studier med legemidler til mennesker, og hvor formålet er å legge til rette for flere kliniske studier, arbeides det dermed for å legge til rette for at flest mulig av kriteriene til legemiddelfirmaene blir møtt, samtidig som studienes kvalitet skal vurderes som god av myndighetene.
12.6.1 Saksbehandlingstid, involverte instanser og høring
Saksbehandlingstider er forskjellige for innesluttet bruk og utsetting av GMO i Norge. Saksbehandling for innesluttet bruk er vanligvis 45 virkedager. Grunnen til forskjellige saksbehandlingstider skyldes flere faktorer. Det er først og fremst at helse- og miljørisikovurdering ved innesluttet bruk er mindre kompleks, da forutsetningen for den innesluttede bruken er at GMO-en ikke skal slippe ut til omgivelser og miljøet. Innesluttet bruk kan i visse tilfeller kun kreve melding, mens en utsetting som hovedregel er søknadspliktig. En annen årsak er at søker i utsettingssøknader også må levere en egenvurdering av bærekraft, samfunnsnytte og etikk. Myndighetene har fått tilbakemelding om at denne egenvurderingen, i tillegg til at alle søknader om utsetting skal ut på høring, oppleves utfordrende for søkere.
Når det gjelder saksbehandlingstider forholder alle instanser seg til saksbehandlingstider fastsatt i EØS-regelverket, og bestreber seg på å behandle søknader så raskt som mulig etter god forvaltningsskikk. Når det gjelder GMO-legemidler har etatene siden 2018 lagt vekt på at det er viktig å legge til rette for samtidig innsendelse av søknader etter alle regelverk for at søker kan oppnå de godkjenninger som den behøver innen ønsket og planlagt oppstart av studien.
I flere EU-land har man sett behov for og gevinst av å formalisere samhandlingen mellom alle berørte regelverk/myndigheter gjennom et koordinerende organ, for eksempel har Nederland et genterapikontor (Gene Therapy Office) som formidler kontakt mellom de ulike myndigheter og søker. Dette kontoret har ikke vedtaksmyndighet, men fungerer som et informasjonspunkt og kan bidra til å koordinere søknadsbehandlingen opp mot søker og mellom myndighetene.
I Sverige og Tyskland fungerer legemiddelmyndigheten som det koordinerende organet (single submission procedure), og mottar søknader under både klinisk utprøving regelverket og GMO-regelverket. Det fattes et felles vedtak etter begge regelverk av legemiddelmyndigheten. Søker forholder seg dermed kun til en myndighet, men myndighetene har kontakt seg imellom.
EUs forordning (EU) nr. 536/2014 om klinisk utprøving av legemidler til mennesker kom til anvendelse i januar 2022. Forordningen innebærer at kliniske utprøvinger godkjennes sentralt i én felles godkjenning for de landene som søker ønsker at studien skal gjennomføres i. Vurdering av GMO-aspekter etter direktiv 2001/18/EF er ikke tatt inn i forordningen, slik at søknader etter GMO-regelverket for kliniske utprøvinger fortsatt må godkjennes nasjonalt, og ikke gjennom sentral prosedyre.
Med overføringen av vedtaksmyndighet til Legemiddelverket, kan løsningen i Norge nå sees på som lignende den svenske og tyske. En forskjell er at det fattes to27 vedtak, ettersom ny forordning for klinisk utprøving ikke legger til rette for ett samlet vedtak. Det samme må gjøres i Sverige og Tyskland. En annen forskjell gjelder innesluttet bruk, det er Helsedirektoratet som er ansvarlig myndighet, og som eventuelt godkjenner innesluttet bruk i en utprøving. Iglesias-Lopez et al. (2019) foreslår noen alternativer for samordning av legemiddelsøknad og GMO-søknad som at GMO-søknaden også kan sendes inn til studielandet gjennom samme EU-søknadsportal, eller at GMO-aspektene integreres i legemiddelsøknaden, og at disse blir godkjent samtidig, som for legemidler til markedsføring. Flere land har uttrykt ønske om en slik samkjøring, deriblant Nederland.
12.6.2 Anbefaling fra utvalget om vedtaksmyndighet og organisering av regelverket
Utvalget vurderer at det er hensiktsmessig med én inngangsport for søker, og at dette er Legemiddelverket. Utvalget har noe delte syn på hvilken etat som bør være vedtaksmyndighet etter genteknologiloven for utsetting. Flertallet (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) går inn for at Legemiddelverket skal være vedtaksmyndighet for klinisk utprøving av GMO-legemidler til både mennesker og dyr. Et mindretall på 4 medlemmer (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) går inn for Legemiddelverket som vedtaksmyndighet for klinisk utprøving av legemidler til mennesker. For klinisk utprøving av legemidler til dyr går de inn for en ordning som innebærer at Miljødirektoratet gis en mulighet til å stanse godkjenningen innen en gitt frist der direktoratet er uenige med Legemiddelverkets miljørisikovurderinger. Ved uenighet foreslår disse utvalgsmedlemmene at uenigheten løftes til de to overordnede departementene.
Flertallet (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) mener at GMO-legemidler i sin helhet bør reguleres i legemiddelregelverket og forvaltes av helsemyndighetene. Myndigheten til å godkjenne søknader om klinisk utprøving av GMO-legemidler etter genteknologilovens §10 ble overført fra Klima- og miljødepartementet til Helse- og omsorgsdepartementet i 2021. Det er videre allerede gjort et unntak for GMO-legemidler som er godkjent for markedsføring etter legemiddelloven slik at de ikke trenger tillatelse til omsetning etter genteknologiloven. Dette unntaket bør komme frem av genteknologiloven. For kliniske studier med GMO-legemidler bør genteknologi-spesifikke regler fremdeles gjelde, i tråd med EØS-regelverk, men flyttes til legemiddelregelverket. Når det gjelder kliniske studier med GMO-legemidler, legger flertallet opp til at dette bør hjemles i legemiddelloven, og at dette bør forvaltes av helsemyndighetene, slik at det blir en helhetlig regulering og praksis både på GMO-legemidler spesielt, men også i sammenheng med legemiddelområdet generelt.
Flertallet mener at forslaget om flytting til legemiddelregelverket bør gjelde kliniske studier både til mennesker og dyr.
Slik opprettholdes en sektororganisering av regelverket på samme måte som foreslått i regelverk for mat og fôr i kapittel 10. Dette er også i tråd med EU-kommisjonen sine forslag til regelverksendringer på legemiddelområdet når det gjelder GMO-legemidler til mennesker.
Flertallet understreker at det materielle innholdet i reguleringen – med krav til vurdering av helse- og miljørisiko for godkjenningspliktige GMO-legemidler – skal beholdes ved en slik flytting.
Flertallet foreslår at legemiddelmyndighetene vurdere nærmere hvordan slik regulering knyttet til GMO-legemidler blir hjemlet i legemiddelloven. Flertallsmedlemmene oppfordrer forvaltningen til straks å igangsette dette arbeidet med å endre regelverket.
Flertallet mener at godkjenning etter GMO-regelverket innlemmes i den sentraliserte prosedyren for godkjenning av kliniske studier i EU/EØS (CTIS – søknadsportalen for kliniske utprøvinger av legemidler til mennesker). Dette flertallet anser at en slik harmonisering og samkjøring vil bidra til å redusere den regulatoriske byrden ved fragmenterte og lite enhetlige prosedyrer i enkeltland, som av flere har blitt pekt på som en av de største hindringene for kliniske studier med GMO-legemidler i Europa. Dette er i tråd med EU-kommisjonens forslag fra april 2023.
Flertallet anbefaler videre at norske beslutningstakere ser hen til EUs forslag til nytt regelverk på legemiddelområdet (pharmaceutical legislation)28 der det blant annet foreslås store endringer i prosessen for vurdering av GMO-legemidler i klinisk utprøving til mennesker. Se 12.1.3 for mer informasjon om de foreslåtte endringene. Målet i EU er å harmonisere regler og praksis mellom landene for å sikre rettferdig og likeverdig tilgang til lovende behandlinger for pasientene samt å tilrettelegge for trygg og effektiv forskning og innovasjon innen avanserte terapier.
Mindretallet (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) mener at GMO-legemidler fremdeles bør reguleres under genteknologiloven. Mindretallet legger vekt på miljøhensyn. En slik løsning harmonerer også best med mindretallets anbefaling om at det skal foretas vurdering av etisk forsvarlighet etter genteknologiloven for legemidler til dyr, og vektleggingen av høring. Mindretallet viser videre til at forslaget forutsetter at EU endrer sitt regelverk, og at slik endring foreløpig ikke er vedtatt av EU.
Utvalget er samstemte i at ved søknader om utsetting ved klinisk utprøving av legemidler til mennesker og dyr, bortsett fra ved forenklet vurdering (melding), skal VKM alltid foreta en risikovurdering, og Miljødirektoratet skal alltid komme med en uttalelse. Vurdering ved klinisk utprøving av legemidler til dyr blir nærmere omtalt i 12.5. Et samlet utvalg presiserer viktigheten av uavhengige risikovurderinger.
Når det gjelder høring av søknader om klinisk utprøving av GMO-legemidler, har utvalget delte meninger om høring skal gjennomføres ved klinisk utprøving av GMO-legemidler til mennesker og ved klinisk utprøving av legemidler til dyr.
For søknader om kliniske studier med GMO-legemidler til utsetting som foreslås å være godkjenningspliktige, anbefaler flertallet på 7 medlemmer (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) at det heller ikke gjennomføres høring, verken ved utprøving til mennesker eller dyr. Dette er i tråd med flertallets anbefaling om at forsøksutsetting av PB- og GMO-organismer til annen bruk ikke bør høres, som beskrevet i kapittel 10. Kliniske studier er også forsøksutsetting.
Flertallet mener at det er tilstrekkelig å legge ut informasjon om søknaden på nettsidene til ansvarlig forvaltningsinstans (Legemiddelverket), i tråd med praksis i Sverige og Tyskland. Videre viser dette flertallet til at det som regel kommer få høringssvar, det vil si 1–4, og anser at hensyn knyttet til åpenhet og informasjon til offentligheten derfor ivaretas tilstrekkelig gjennom foreslåtte informasjonsordning. Flertallet mener at det ikke er noe prinsipielt problematisk ved GMO-legemidler sammenlignet med andre legemidler, og at relevante hensyn som bør ligge til grunn for vurdering av søknader om kliniske studier er av faglig karakter. Dette mener flertallet best ivaretas av kompetente myndigheter og ikke offentligheten ellers. Flertallet viser videre til at en slik endring forenkler saksbehandlingen og korter ned tiden det tar å få en godkjenning etter genteknologiregelverket på plass. Dette vil både frigjøre ressurser i forvaltningen som kan brukes på andre oppgaver samt bidra til å gjøre Norge mer attraktivt for kliniske studier med genterapi og andre GMO-legemidler fordi prosessen vil være mindre krevende for søker.
Forslaget om å unnlate høring for GMO-legemidler, både til forenklet utsetting (melding) og godkjenningspliktige søknader om utsetting, vil sannsynligvis kreve lovendring.
Et mindretall på 4 medlemmer (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) går inn for et fortsatt krav om å ha høring for søknader om utsetting som angår klinisk utprøving av GMO-legemidler til mennesker og til dyr (både meldinger om forenklet utsetting og øvrige søknader om utsetting). Dette mindretallet anser at et fortsatt krav om høring for søknader om utsetting bidrar til åpenhet, tillit og saksopplysning, og at høring enkelt gjennomføres digitalt innenfor avsatt saksbehandlingstid. Dette er i tråd med formålet til åpenhetsforordningen som nylig har trådt i kraft i EU, og prinsippet om meroffentlighet i norsk forvaltning. Basert på argumentene over, vurderer dette mindretallet av utvalget at nytten av en høring for utsettingssøknader veier tyngre enn at søker eventuelt kan oppleve høring som belastende.
12.6.3 Innesluttet bruk og utsetting
12.6.3.1 Vurdering kun som innesluttet bruk
Tre land i EU opplyser å godkjenne GMO i legemiddelutprøving kun etter regler om innesluttet bruk. Fordeler med en slik modell kan være at man forholder seg bare til en GMO-myndighet. Innesluttet bruk regelverket er satt sammen slik at dersom studiestedet har godkjente fasiliteter for arbeid med GMO på riktig inneslutningsnivå for GMO-legemidlene som skal prøves ut, behøves kun en melding for hver nye GMO, uavhengig av GMM-ens risikonivå. Det vil si at dersom man tidligere har fått godkjent lokalet for bruk på et bestemt nivå 1 – 4, og skal utføre ny studie, da trenger man ikke søknad, men melding. En slik melding vurderes ofte raskt, fordi myndighetene i utgangspunktet ikke krever en omfattende helse- og miljørisikovurdering, og GMO-en skal arbeides med i et lukket system uten utslipp til omgivelsene.
Legemiddelindustrien, en rekke pasientforeninger og kliniske fagmiljøer ved sykehusene har i sine høringssvar uttrykt et ønske om at vurdering som innesluttet bruk benyttes i stor grad. De legger vekt på at melding om innesluttet bruk er mindre omfattende, tar kort tid å behandle og er hensiktsmessig når et legemiddel ikke kommer i kontakt med miljøet eller kun har neglisjerbar miljørisiko. Videre fremhever de at en slik forenklet prosess vil gjøre Norge til et mer attraktivt land å legge kliniske studier med GMO-legemidler til, i tråd med handlingsplanen for kliniske studier, og vil komme både pasienter og offentlige og private helseaktører til gode. To av tre land i EU som godkjenner legemiddelutprøvinger som innesluttet bruk, gjennomfører heller ikke høring av søknaden. Dette er i tråd med norsk regelverk om innesluttet bruk. Helsedirektoratet har vurdert at krav til innesluttet bruk ved utprøving av humane genmodifiserte celler etter genteknologiloven er for strenge, og ikke formålstjenlige. For slike studier vil også forskrift om håndtering av humane celler og vev komme til anvendelse.
12.6.3.2 Vurdering kun som utsetting
Elleve land opplyser å godkjenne kliniske utprøvinger kun som utsetting, ti land som både innesluttet bruk og utsetting og tre land som innesluttet bruk. Av de landene som godkjenner kun som utsetting, er det ikke klart ut ifra den offentlig tilgjengelige informasjon på EUs nettsider, om disse landene praktiserer at utsettingen også innbefatter mottak, lagring og tilberedning og enkel istandgjøring av GMO-legemidlet på studiestedet, eller om disse aktivitetene ikke anses å være en del av den kliniske utprøvingen29.
Norske myndigheter har etablert en praksis hvor innesluttet bruk-forskriften kommer til anvendelse for mottak, lagring og håndtering av GMO-legemidlene som må ha inneslutningstiltak på nivå 1 eller 2 avhengig av GMO-en. De fleste GMO-legemidler til mennesker vil håndteres på nivå 1 eller 2. Ved godkjenning av innesluttet bruk skjer godkjenning av hvert lokale/rom på studiestedet/stedet hvor GMO-en skal mottas/lagres/håndteres. Dersom man allerede har slike godkjente fasiliteter som er tenkt brukt er det kun behov for melding om bruk av GMO-legemidlet. Erfaringsmessig har mange sponsorer (ansvarlig for utprøvingen) ikke slike fasiliteter godkjent fra før i Norge. Da må det søkes om godkjenning av fasiliteten før bruk.
Dersom man skulle velge å se bort fra innesluttet bruk-regelverket, og godkjenne alle prosesser kun som utsetting, vil søkere kun måtte forholde seg til eventuelle risikohåndteringskrav30, (for eksempel at avfallsbehandling for GMO-en skal skje på bestemte måter) gitt i ett vedtak om utsetting. Det ville ikke ha vært behov for å ha fasiliteter som oppfyller kravene til innesluttet bruk, og i et slik alternativ må søker bare forholde seg til én myndighet (vedtaksmyndigheten). Dette kan oppleves forenklende for søker. Modellen samsvarer også godt med modellen for søknader om markedsføringstillatelse, hvor det også kun skjer en godkjenning av legemiddelmyndighetene, men hvor GMO-myndighetene spiller inn om miljørisiko og miljørisikohåndtering i en egen GMO-prosedyre for hver søknad. Samtidig er det forskjeller i behov for håndtering i en klinisk utprøving og et legemiddel som skal ut på markedet (se omtale av datagrunnlag i 12.5.4).
Eventuelle ulemper med denne modellen kan være dersom GMO-en har egenskaper som tilsier at man burde arbeidet med denne under innesluttede forhold, for å hindre unødig spredning og fare for omgivelsene ved uhell. Dette gjelder for eksempel når man mottar legemidlet som en konsentrert virus-batch, som må åpnes og fortynnes i en væske til intravenøs pose/sprøyter eller annen tilvirking før det kan gis i riktig dose til pasientene i utprøvingen. Dette er gjerne hovedregelen for GMO-legemidler med adeno-assosierte virusvektorer. Det bør vurderes om eventuell risiko er av en slik art at man ved mottak, lagring og håndtering av GMO som er ment til legemiddel, kan se bort fra sikkerhetshensynene i innesluttet bruk-forskriften, og eventuelt godkjenne dette i en godkjenning av utsetting. Videre kan det være at godkjenning kun som utsetting, kan være en hensiktsmessig løsning kun for spesifikke typer GMO-legemidler. Eksempel på slike GMO-legemidler, er ikke-replikerende virusvektor-vaksiner som kommer ferdig til bruk (med lavere antatt miljørisiko sammenlignet med for eksempel legemidler som er konstruert med replikerende virusvektorer). Avhengig av GMO-ens egenskaper, kan tillatelsen til utsetting komme med spesifikke vilkår for å håndtere risiko, dersom miljørisikovurderingen tilsier det.
Det vil være viktig å sikre at den kliniske utprøvingen ikke medfører noen risiko for andre, sårbare pasienter på sykehuset. Dersom utprøvingen ikke skjer i lokaler som er godkjent for innesluttet bruk, kan det settes vilkår i tillatelsen til utsetting for å ivareta dette. Dette kan for eksempel være vilkår knyttet til avtrekk, rengjøring, eget toalett, hvordan pasienten forlater sykehuset osv. Utvalget anbefaler at helsemyndighetene setter opp en liste med standardvilkår for håndtering av GMO-legemidler som ikke krever innesluttet-bruk fasiliteter, for å sikre korrekt risikohåndtering på sykehus og behandlingslokaler, og for at det skal være tydeligere for søkere hvilke krav som stilles.
12.6.3.3 Forhåndsvurdering av om søknaden innebærer utsetting
Fra 2018 gikk etatene i stor grad bort fra modellen om å forhåndsvurdere om studien medførte utsetting, grunnet ny juridisk vurdering fra Helsedirektoratet. Klinisk utprøving av GMO-legemidler godkjennes etter denne fortolkningen kun som innesluttet bruk i sin helhet dersom hele studien gjennomføres i fasiliteter godkjent for innesluttet bruk. Kun i tilfeller med terminale pasienter på egen avdeling/i eget rom, kunne det vært aktuelt at hele utprøvingen skulle ha gått som innesluttet bruk i sin helhet. Dersom pasient forlater studiestedet er dette ansett som utsetting av GMO (gjennom pasient). Ifølge Anliker et al. (2010), ser det ut til å være vanlig å vurdere om studien innebærer utsetting av GMO eller ikke basert på studieoppsettet.
En fordel med en slik juridisk avklaring mellom etatene er at den gir tydelighet og forutsigbarhet for søker. Man vurderer ikke i hver enkelt sak om studien medfører utsetting eller ikke, og søker kan dermed søke godkjenning etter regelverk om innesluttet bruk og utsetting samtidig. Det har også medført mindre behov for avklaringer mellom GMO-myndighetene, og mellom myndighetene og søker, for å avgjøre hva som gjelder for den enkelte studie, hvilket dokumentasjonskrav som gjelder, og om studien medfører utsetting eller ikke. En sekvensiell behandling vil kunne være forsinkende for søker sammenlignet med en samtidig innsendelse av søknader til begge myndigheter.
Samtidig kan et slikt system muligens oppfattes mer rigid av søkerne, dersom det kan bevises at GMO-en ikke frigis fra forsøkspersonene etter den er gitt, eller at de ikke er påvisbare etter en viss tid (kort tidsrom på studiestedet). Utprøvingen blir dermed i praksis ikke utsetting av GMO. Det kan være en mer robust forvaltning av GMO-legemiddelområdet å gjøre en forhåndsvurdering av om GMO i legemiddelutprøving fører til utsetting. Flere EU-land opplyser at kliniske utprøvinger med GMO-legemidler godkjennes etter både regelverket om innesluttet bruk og utsetting.
En problemstilling i forbindelse med en slik forhåndsvurdering av om studien innebærer utsetting av GMO til omgivelsene eller ikke, er at en slik vurdering i noen tilfeller vil være basert på foreløpige resultater og begrenset informasjon, fordi selve studien vil gi informasjon som er relevant for å gjøre nettopp denne vurderingen. Det kan dermed være vanskelig å konkludere på forhånd om studien vil innebære utsetting eller ikke. GMO-regelverket er også tydelig på at alt som ikke er innesluttet bruk er utsetting, og gir ikke noen andre kriterier for å vurdere om noe blir utsetting.
12.6.4 Biologisk inneslutning – forenklet utsetting?
Et konsept i denne sammenheng er biologisk innesluttethet eller biologisk inneslutning. Man kan tenke seg at når man gir et genmodifisert virus eller modifiserte celler til et menneske, så vil noen ha egenskaper som gjør at de er å anse som biologisk innesluttet, hvor kroppen fungerer som et slags biologisk lukket system så lenge GMO-legemiddelet ikke overlever utenfor. En slik kategori brukes blant annet i Storbritannia31.Innesluttet bruk er etter genteknologiloven i dag beskrevet som at arbeid med GMO skal skje i et lukket system med fysiske inneslutningstiltak, og at innesluttet bruk skal skje i godkjente laboratorier og anlegg. Biologisk inneslutning innebærer at det er lite sannsynlig at et legemiddel kommer ut av kroppen og/eller utgjør en neglisjerbar miljørisiko om det gjør det.
Et eksempel er humane genmodifiserte celler, som ikke vil overleve utenfor en kropp og det kan derfor anføres at cellene kan anses som biologisk innesluttede i et menneske. Etter at slike celler er satt inn i en pasient/studiedeltager, er det ikke lenger cellene som er omfattet av genteknologiloven32, men den genmodifiserte virusvektoren som eventuelt er til stede i cellene. Avhengig av egenskapene og framstillingen av den genmodifiserte virusvektoren, kan denne virusvektoren potensielt ha mulighet til å spre seg ut fra testpersonen og til miljøet, og eventuelt i ytterste konsekvens gi opphav til et nytt virus. Dette undersøkes og gjennomgås i en miljørisikovurdering.
Det kan være utfordrende å bekrefte fullstendig fravær av for eksempelvis virusvektorer, men det er mulig å dokumentere et neglisjerbart nivå. Dersom søker kan dokumentere et neglisjerbart nivå av resterende viruspartikler i cellene/legemidlet, og i tillegg dokumentere fravær av replikerende virusvektorer33, kan også miljørisiko anses å være neglisjerbar. For bestemte typer humane genmodifiserte celler og visse virusvektorer er det faglig enighet i EU/EØS om at disse kan vurderes til å ha neglisjerbar miljørisiko. Dersom det kan dokumenteres at det er høyst usannsynlig at den genmodifiserte virusvektoren kan spres ut fra testpersonen i miljøet, eller ikke utgjør en betydningsfull miljørisiko om den spres, altså at miljørisikoen er neglisjerbar, kan man lage et system for forenklet saksbehandling. En slik forenklet saksbehandling kan raskere iverksettes ved å vurdere søknader som forenklet utsetting fremfor å vurdere søknaden som biologisk innesluttet, fordi sistnevnte krever lovendring for å utvide definisjonen av innesluttet. Slik saksbehandling kan gjelde for GMO-legemidler som det er enighet i EU-gruppene (se 12.1.3 og 12.2.3) om at har neglisjerbar risiko. Alle GMO-legemidler som kvalifiserer for skjemaer med en ferdig utfylt miljørisikovurdering (ERA) bør kunne behandles som forenklet utsetting.
Anbefalinger fra utvalget
Utvalgets mandat omfatter blant annet regler for innesluttet bruk av GMO, noe som setter viktige rammer for utvikling og bruk av GMO-legemidler. Dagens regler og praksis utgjør betydelige flaskehalser for skalering av behandling med gen- og celleterapi for alvorlig syke pasienter, blant annet i form av særkrav til oppbevaring av biologisk materiale, utforming av infrastruktur med mer. Dette gjelder ikke bare kliniske studier, som behandlet i denne NOUen, men også annen innesluttet bruk både til forskning og innovasjon, produksjon av legemidler, samt bruk av godkjente legemidler i den offentlige helsetjenesten. Grunnet kapasitetsbegrensninger i utvalgsarbeidet anbefaler flertallet (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) at regler for innesluttet bruk av GMO gjennomgås i en egen prosess i oppfølgingen av denne NOUen. En slik gjennomgang bør delegeres til en dedikert ekspertgruppe (ikke et offentlig utvalg) satt sammen av fagfolk og representanter fra forvaltningen som har god kjennskap til innesluttet bruk av GMO, særlig GMO-legemidler.
Flertallet mener subsidiært, dersom ikke bestemmelsene om GMO-legemidler i sin helhet skal forvaltes av helsemyndighetene, at det heller opprettes en kategori biologisk innesluttet for legemidler som kvalifiserer for EU-skjema. Slik vil intensjonen i forslaget om forenklet utsetting beholdes (et differensiert system som åpner for melding for legemidler med forutsigbar lav/neglisjerbar risiko), samtidig som det sikres at bestemmelsene forvaltes av helsemyndighetene, siden disse har ansvaret for regler om innesluttet bruk. Innføring av en ny slik kategori krever lovendring. Flertallet ber om konkrete innspill på valg mellom klassifisering forenklet utsetting og biologisk inneslutting fra relevante aktører i høringen som følger NOUen.
12.6.4.1 Humane genmodifiserte celler – klargjøring av avgrensinger
Som nevnt har søknader om klinisk utprøving av humane genmodifiserte celler allerede en forenklet saksbehandling, og det er avklart i EU/EØS at humane genmodifiserte celler i kultur er å anse som en genmodifisert organisme. At mennesket aldri kan bli en genmodifisert organisme etter loven er relevant å nevne i denne sammenheng, dette presiseres tydelig i forarbeidene; definisjonen (av genmodifiserte organismer) omfatter ikke mennesker.
Utvalget anser imidlertid at det bør fremgå klarere av loven at definisjonen av genmodifiserte organismer ikke omfatter mennesker på noe trinn i utviklingen fra befruktet egg til fullt utviklet individ, men at den omfatter humane celler i kultur. Videre, når det gjelder humane genmodifiserte celler som settes inn i mennesket igjen, har praksis vært at genteknologiloven da ikke omfatter cellene, men den genmodifiserte virusvektoren som eventuelt måtte finnes i cellene. Det kan være klargjørende at dette fremgår av regelverket.
12.6.4.2 Krav til forenklet utsetting
For at et nytt grep som forenklet utsetting skal fungere i praksis, må det spesifiseres tydelige krav som skal oppfylles. Her anser utvalget at det er hensiktsmessig å se hen til de felles søknadskjemaene som er utarbeidet i EU, og som Norge har sluttet seg til. Søknadskjemaer som Norge allerede har tilsluttet seg, for eksempel skjema for humane genmodifiserte celler, og søknadskjema for adeno-assosierte virusvektorer34 kan fungere som et terskelnivå for at klinisk utprøving av GMO-legemidler til mennesker kan behandles etter lempeligere regler som blant annet omfatter meldeplikt i stedet for søknadsplikt. For å kunne vise til en generell miljørisikovurdering i vedlegget i skjemaet, forutsettes at søker kan dokumentere at vilkår for neglisjerbar miljørisiko er oppfylt, for eksempel at eventuelle resterende virusvektor-partikler er på et neglisjerbart nivå. I fremtiden vil andre vilkår kunne legges til grunn for nye skjemaer, for eksempel at GMOen har egenskaper som gjør at den ikke overlever utenfor mottaker.
Utvalget anser at dette er en pragmatisk løsning for krav til forenklet utsetting. Ved utarbeidelse av flere slike omforente søknadskjema, hvor bruken av disse forutsetter at søker kan dokumentere at vilkår for en slik neglisjerbar miljørisiko er oppfylt, kan det utvide hvilke GMO-legemidler som kan anses å føre til biologisk inneslutning, altså kunne behandles som forenklet utsetting. Utvalget mener det skal foretas en målrettet høring ved utarbeidelse av nye skjema.
For å sikre at utprøving av slike GMO-legemidler, som ikke vil kreve innesluttet bruk-fasiliteter, skjer på en sikker måte, anser utvalget at det bør settes spesifikke vilkår i myndighetenes svar på meldingen, til hvordan GMO-legemidlene skal håndteres. Dette gjelder spesielt adeno-assosierte virusvektorer, som kan overleve en viss periode på overflater slik som laboratoriebenker. Dette tilsier at ordningen med melding bør utformes slik at myndighetene kan stille vilkår, og slik at virksomheten ikke kan starte den kliniske utprøvingen før svar på meldingen er mottatt.
Videre anser utvalget at bestemmelser om forenklet utsetting bør komme frem av forskrift, da de forutsetter et detaljnivå som hører hjemme der, og at GMO-legemidler som kan anses å føre til neglisjerbar risiko ikke skal være søknadspliktig etter genteknologiloven. En melding anses tilstrekkelig. Utvalget anser at genteknologiloven § 10 (om godkjenning av utsetting) femte ledd antagelig vil gi tilstrekkelig hjemmel til dette. Femte ledd gir anledning til å gi forskrift om at det kun kan kreves melding for bestemte typer GMO som settes ut i bestemte miljøer, og det antas at forskriftshjemmelen også kan omfatte bruk av GMO-legemidler i kliniske utprøvinger der dette kan anses å føre til biologisk inneslutning. Høring vil antakelig også kunne unnlates. Paragraf 13 annet punktum slår fast at det alltid skal gjennomføres offentlig høring i saker som gjelder godkjenning av søknad om utsetting av genmodifiserte organismer. Her vil det imidlertid ikke være noen søknad som skal godkjennes, men en melding som skal sendes til myndighetene, og kravet om høring kommer dermed i utgangspunktet ikke til anvendelse. Dersom saken anses som et tiltak med vesentlige virkninger, skal forslaget normalt legges ut på høring etter utredningsinstruksen § 3-3 første ledd. Høring kan unnlates på visse vilkår. Det antas at vilkåret om vesentlige virkninger vanligvis ikke vil være oppfylt i disse sakene. Dersom også andre krav skal fravikes, kan en ny lovhjemmel være nødvendig.
Utvalget anser at bruk av forenklet utsetting i regelverket i Norge ikke er i strid med EUs praksis på området. EU lar det være opp til landene å bestemme om klinisk utprøving med GMO-legemidler skal behandles etter regelverk om innesluttet bruk eller utsetting.
12.6.5 Risikovurdering
For utsetting av GMO i miljøet, er det eventuelle skadelige konsekvenser av utsettingen som skal vurderes, og det skal foreslås tiltak for håndtering og kontroll av eventuell påvist risiko. Vitenskapelige og uavhengige risikovurderinger etter genteknologiloven av GMO til utsetting er et viktig prinsipp for myndighetene. Flere andre EU-land opplyser å benytte vitenskapelige miljøer i risikovurderingen av søknader om GMO-legemidler, dette er derfor ikke uvanlig i EU-sammenheng.
Klinisk utprøving er forskning35 hvor man skal innhente data som sier noe om sikkerheten og effekten ved bruken av GMO-en. En klinisk utprøving av et legemiddel har til hensikt å skaffe til veie, eller etterprøve informasjon om dets effekt og sikkerhet. Etter GMO-regelverket er det spesielt eventuell risiko for spredning av GMO-en (GMO-legemidlet) som har betydning, og et mål på dette er hvor mye frigivelse (shedding) av den genmodifiserte virusvektoren man har fra pasienten. Viktigheten av at kliniske utprøvinger dokumenterer shedding av GMO-en fra testsubjektene trekkes frem av Anliker et al. (2010), da en slik frigivelse av GMO til omgivelser (andre mennesker, dyr) og/eller miljøet (via avløp, annet) er viktig for å kunne vurdere miljørisiko av GMO-en senere, på markedsføringsstadiet. Forfatterne foreslår også en forenklet inndeling av ulike GMO i legemidler og en risiko for utskillelse per gruppe som en pekepinn.
Type GMO i legemidlet | Eksempler | Risiko for utskillelse |
|---|---|---|
Genmodifiserte celler | Neglisjerbar | |
Ikke-replikerende virusvektorer | Lav | |
Genmodifiserte virus | Høy | |
Genmodifiserte bakterier | Høy |
Utviklingen av GMO-legemidler er i rask progresjon, og metodene for molekylære og cellulære teknikker utvikles stadig. I likhet med genmodifisering av planter og dyr, er det også vitenskapelige usikkerheter om disse genmodifiserte mikroorganismene som utvikles til bruk som legemidler, både om virkning og utilsiktede effekter.
Med GMO-interplay arbeidet og utvikling av søknadsskjemaer og tilhørende veiledningsdokumenter, har myndighetene forsøkt å gjøre en slik type inndeling av GMO-legemidler, og tilpasset omfang av og krav til risikovurderingen deretter. Det gjøres imidlertid fortsatt en sak-til-sak-vurdering av risiko, som er et viktig prinsipp i GMO-regelverket.
12.6.6 Genteknologilovens kriterier om bærekraft, samfunnsnytte og etikk (BSE)
Det er til dels overlappende krav til vurderinger av samfunnsnytte og etikk i legemiddelregelverket og genteknologiloven. Legemidler som består av eller inneholder genmodifiserte organismer vil kunne brukes til behandling av sykdommer som tradisjonelle legemidler ikke er i stand til. De vil i så måte langt på vei kunne sies å være samfunnsnyttige og etisk forsvarlige.
Disse vurderingene ble lagt til grunn i KLDs lovendringsforslag, som innebærer at disse kriteriene i genteknologiloven ikke skulle gjelde ved behandling av søknad om klinisk utprøving av GMO-legemidler. Høringsinstansene var delt i sitt syn. Av dem som gikk mot forslaget, mente de fleste at lovendringen var problematisk for legemidler til veterinær bruk. Høringsinstansene la blant annet vekt på at regelverket for veterinærmedisin ikke stiller de samme krav til vurderinger av utprøving av legemidler på dyr, som det som gjelder utprøving av legemidler på mennesker. Det vises også til manglende bærekraftvurderinger etter legemiddelregelverket.
Fra humanmedisinsk hold har det vært uttrykt at et særnorsk vilkår for søker med hensyn til å dokumentere genteknologilovens tilleggskriterier er utfordrende, og dette kravet kan dermed anses å redusere innovasjon og utprøving av GMO-legemidler i Norge, noe som kan være uheldig for alvorlig syke mennesker i Norge.
Utvalget anser at det kan være andre hensyn som er relevante når det gjelder GMO-legemidler til dyr, sammenlignet med GMO-legemidler til mennesker, og at det derfor kan være argumenter for å beholde disse kriteriene for veterinære legemidler.
Et unntak vil være forenklende for både søker og myndigheter, og kan bidra til at søkere i større grad tar i bruk genteknologi som kan vurderes som et bedre alternativ til teknologier som ikke reguleres like strengt. Utvalget legger imidlertid vekt på at mens annet regelverk ivaretar relevante hensyn når det gjelder vurdering av samfunnsnytte og etikk for veterinære GMO-legemidler, er hensynet til bærekraft i liten grad dekket.
Bærekraft er etter utvalgets syn et svært viktig vurderingstema. BSE-kriteriene i genteknologiloven omfatter allerede bærekraft i lovteksten, og dette er dekket av tilpasningsteksten til EØS-avtalen. Tilgrensende regelverk for veterinærmedisin har ikke krav om vurdering av bærekraft. Det har videre i Norge vært politisk enighet om et restriktivt GMO-regelverk, og en eventuell oppmykning av reguleringen på et omstridt saksområde kan føre til reduksjon av tillit til myndighetene, i hvert fall i deler av befolkningen.
Flertallet (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) anbefaler at det ikke skal være krav om etikkvurderinger etter genteknologiregelverket på noen kliniske studier til verken mennesker eller dyr (jamfør kapittel 10.2).
Mindretallet (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) mener at BSE-kriteriene ikke skal gjelde for klinisk utprøving av GMO-legemidler til mennesker, men at de bør gjelde for klinisk utprøving av GMO-legemidler til dyr. Samtidig vil mindretallet understreke at samfunnsnytte, bærekraft og etikk ikke kun er relevante kriterier for veterinære legemidler som inneholder GMO, men for alle veterinære legemidler. Utvalgets mandat begrenses imidlertid til GMO-legemidler og ikke legemidler for øvrig.
12.6.7 Suksessfaktorer for høyere antall kliniske utprøvinger med GMO-legemidler i EU-landene
Det er mange faktorer som påvirker hvilke land som velges ut for å gjennomføre en klinisk utprøving. For denne utredningen som gjelder genteknologiloven, og omfatter en vurdering av praksis for klinisk utprøving av GMO-legemidler, er det hensiktsmessig å se nærmere på valgte forvaltningsmodeller i EU/EØS.
Det ser ikke ut til at det er en bestemt forvaltningsmodell som er avgjørende for om landene har mange kliniske studier med GMO-legemidler eller ikke. Når det gjelder studier som godkjennes som utsetting av GMO-legemidler i henhold til del B i utsettingsdirektivet, er det en egen EU-database hvor slike studier offentliggjøres36. Databasen gir kun en oversikt over de tilfeller hvor man har vurdert søknaden som utsetting, og ikke kun som innesluttet. Tall på antall kliniske utprøvinger i land som opplyser at de godkjenner som innesluttet bruk og utsetting (som Norge) er dermed ikke tilgjengelig. Det er derfor vanskelig å sammenligne åpent tilgjengelig data om antall kliniske studier basert på modellene kun utsetting vs. modellen med innesluttet bruk og utsetting.
Det man derimot kan sammenligne, er om det er registrert flere studier i land som opplyser å ha en single submission procedure37, enn i land hvor det er GMO-myndighetene som godkjenner GMO-søknader om utsetting. Av data registrert i utsettingsdatabasen fra 2003 til 2021 er det registrert 193 utsettingsstudier i land hvor legemiddelmyndigheten godkjenner utsetting etter GMO-regelverket. I samme periode er det registrert 372 utsettingsstudier i land hvor søknader sendes GMO-myndighetene for godkjenning av utsetting. Hittil i 2022 er det registrert 15 studier i databasen fordelt på seks ulike studieland, hvor fire av landene er land hvor GMO-myndigheten har ansvaret for å godkjenne studien etter GMO-regelverket (Spania, Italia, Portugal og Irland).
Det kan derfor vanskelig konkluderes med at en single submission procedure alene gir det høyeste antallet kliniske studier i et gitt studieland i EU, men at land som til nå har lang erfaring i å gjennomføre kliniske studier med GMO-legemidler gjerne har flere suksessfaktorer på plass når det gjelder gjennomføring av kliniske studier med GMO-legemidler. En studie fra 2016 (De Wilde et al. 2016), som har gjort en bred gjennomgang av kliniske studier med gen- og celleterapi i Europa, fant blant annet at majoriteten av tidligfasestudier er finansiert av akademia, hvor forskningen og utviklingen finner sted. Dette kan tyde på at det også er viktig å legge til rette for sterke akademiske fagmiljøer som forsker på legemidler. Blant annet fant studien at egne finansieringsordninger for forskning og produksjon av gen- og celleterapier i land som Spania og Storbritannia kunne være viktige kriterier for at disse landene hadde et særlig høyt antall av kliniske studier på GMO-legemidler.
Selv om det ikke kan konkluderes om betydningen av enkelte forvaltningsmodeller på suksessraten, er det bred enighet om at det europeiske GMO-regelverket og forvaltningspraksisen i seg selv har betydning for hvor mange kliniske studier med GMO-legemidler Europa som helhet klarer å tiltrekke seg. EU-kommisjonen skriver i sin Pharmaceutical Strategy at de fragmenterte nasjonale godkjenningsprosedyrene etter GMO-regelverket er til hinder for gjennomføringen av kliniske GMO-studier i Europa. Videre trekker Europaparlamentet frem at erfaringene viser at for kliniske studier med GMO-legemidler er prosessen knyttet til miljørisikovurderingen kompleks og kan ta svært lang tid. En kartlegging gjort av legemiddelindustriens bransjeforeninger i Europa viser at antallet studier med avanserte terapier økte med under 2 prosent i Europa i perioden 2014-2019, sammenlignet med en økning på hhv 28 og 36 prosent i Kina og USA. Produsentene peker på det europeiske GMO-regelverket som en hindring.
12.7 Beskrivelse og vurdering av utvalgets foreslåtte modell for klinisk utprøving GMO-legemidler
Utvalget har i sin behandling av mandatpunktet vurdere norsk praksis for vurdering og godkjenning av GMO-legemiddel til klinisk utprøving, samanlikne med EU sin praksis, eventuelt foreslå endringar hatt en bred diskusjon rundt praksis på området og vurdert alternative modeller for godkjenning av søknader om klinisk utprøving med GMO-legemidler til mennesker og dyr.
12.7.1 Hvorfor foreslås en ny modell og hva er bakgrunnen for forslaget?
Utvalget merker seg at det er få søknader om kliniske studier med GMO-legemidler til mennesker i Norge. For GMO-legemidler til dyr har det ikke vært søkt om kliniske utprøvinger i Norge. Imidlertid ser utvalget at det er et klart problem at syke mennesker i Norge ikke får deltatt i kliniske studier, gjerne der hvor pasientene ikke har et annet reelt alternativ til behandling. Noen saker har vært oppe i media, hvor forvaltningens befatning med søknadsbehandling har blitt omtalt og kritisert for å være utslagsgivende for Norges mulighet til å gjennomføre studiene. Både søkere og myndigheter har sett behov for avklaring av praksis, og at regelverket kan tydeliggjøres på visse punkter.
Et klart formål med å inkludere dette punktet i utvalgets mandat, er å oppnå en enklere og mer effektiv saksbehandling, og mindre uklarheter i relevant regelverk. Utvalget tilslutter seg ønsket om flere kliniske studier til Norge. Dette kan ses i sammenheng med legemiddelstrategi-arbeidet som pågår både i EU og i Norge, og utvalgets anbefalinger vil legge til rette for at vi har en harmonisert praksis med EU på området. Tiltakene er også i samsvar med tiltakene i regjeringens nasjonale handlingsplan for kliniske studier der et av innsatsområdene er å gi pasienter økte muligheter for å delta i kliniske studier.
Utvalgets beskrivelse og vurdering av praksis ble gjennomgått i 12.6, og utvalget er langt på vei enige om godkjenningsmodell. Det presenteres alternativer når det gjelder hvilken etat utvalget vurderer bør være vedtaksmyndighet, og om høring for søknader om utsetting skal gjennomføres. Mindretallet anser at bærekraft, samfunnsnytte og etikk fortsatt bør vurderes for GMO-legemidler til dyr. Utvalget foreslår kravet sløyfet for utprøving av GMO-legemidler til mennesker ettersom samfunnsnytte og etikk ivaretas av andre (etikk-komite og Legemiddelverket) når det gjelder kliniske legemiddelutprøvinger.
12.7.2 Forslag til ny modell for vurdering etter genteknologiloven
Under oppsummeres punktvis nytt forslag til modell for godkjenning av søknader om kliniske studier med GMO-legemidler etter genteknologiloven til mennesker og dyr, og lenger ned visualisert i figur 12.3:
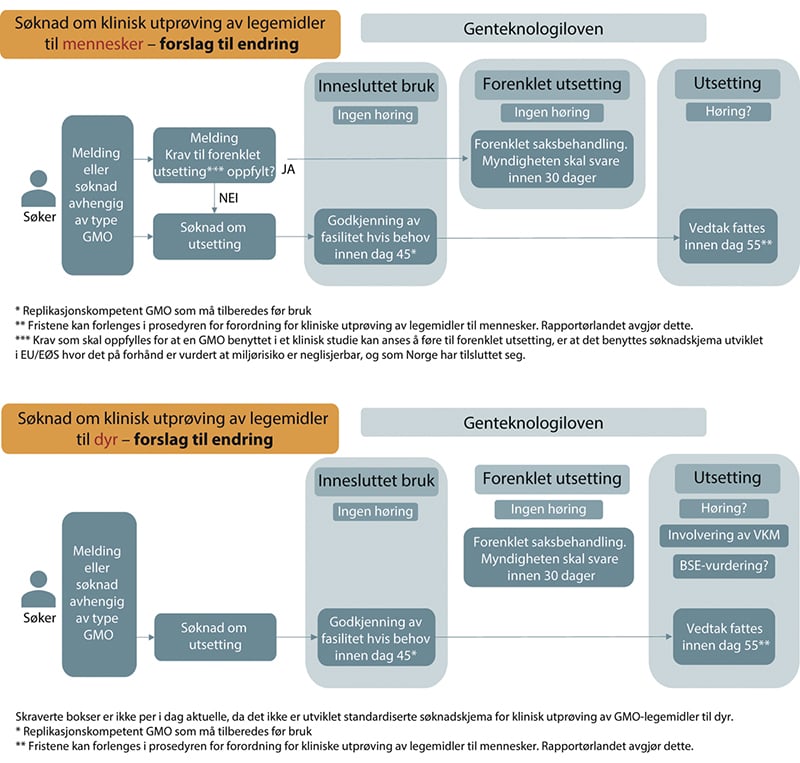
Figur 12.3 Skjematisk oversikt over forslag til saksgang
Første instans og alternativ vedtaksmyndighet
Legemiddelverket er inngangsport for søkere, og Legemiddelverket koordinerer saksbehandlingen mellom involverte etater; Legemiddelverket, Helsedirektoratet og Miljødirektoratet. Foreslått vedtaksmyndighet varierer mellom alternativene. Tider for saksbehandling etter genteknologiloven er tilpasset godkjenningsprosedyrene for klinisk utprøving etter legemiddelregelverk.
Innesluttet bruk
Både regelverk om innesluttet bruk og utsetting benyttes i revidert modell, men innesluttet bruk-regelverket i mindre grad enn for dagens praksis.
Regulering av kliniske utprøvinger hos mennesker med humane genmodifiserte celler og andre GMOer med dokumentert neglisjerbar miljørisiko etter genteknologiloven
Reguleres som forenklet utsetting der kravene er tilfredsstilt. Her kreves kun melding til Legemiddelverket. Dersom krav til forenklet utsetting ikke er oppfylt i meldingen, må søker ettersende dokumentasjon og status endres eventuelt til søknad om utsetting.
Vurdering av bærekraft, samfunnsnytte og etikk (BSE)
Hele utvalget mener at det ikke skal foretas en vurdering av BSE for klinisk utprøving av legemidler til mennesker. Ved kliniske studier på mennesker ivaretas etikkvurderinger av REK-KULMU, uavhengig av typen legemiddel.
Når det gjelder klinisk utprøving av legemidler til dyr er utvalget delt. Flertallet mener at det heller ikke skal foretas vurdering av BSE ved klinisk utprøving av legemidler til dyr. Mindretallet mener at fortsatt skal foretas en vurdering av BSE. For nærmere begrunnelser, se 12.7.5.5.
Høring
Utvalget har delte meninger om høring skal gjennomføres ved søknader om utsetting. Mange søknader vil med foreslått modell behandles som forenklet utsetting, og vil da ikke kreve høring. Et flertall på 7 medlemmer (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) mener at høring bør utgå for alle kliniske utprøvinger til både mennesker og dyr. Et mindretall på 4 medlemmer (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) ønsker å beholde høring.
Bruk av søknadskjema
Alle EUs ferdigutviklede søknadsskjemaer kan benyttes, og alle GMO-legemidler som kvalifiserer for skjema kan ha en forenklet behandling hos vedtaksmyndigheten. I praksis innebærer dette en melding til myndighetene.
12.7.3 Første instans og alternativ vedtaksmyndighet – fordeler og ulemper
Revidert modell har to alternativ når det gjelder vedtaksmyndighet for søknader om utsetting av GMO i legemidler i klinisk utprøving, Legemiddelverket alene eller Legemiddelverket og Miljødirektoratet sammen. Se nærmere detaljer om voteringer for utvalgsmedlemmene i 12.6. Et flertall av utvalgsmedlemmene stiller seg bak forslaget om Legemiddelverket som vedtaksmyndighet for klinisk utprøving av GMO-legemidler til både mennesker og dyr etter genteknologiloven. Et mindretall går inn for Legemiddelverket som vedtaksmyndighet for klinisk utprøving av legemidler til mennesker. For klinisk utprøving av legemidler til dyr går de inn for en ordning som innebærer at Miljødirektoratet gis en mulighet til stanse godkjenningen innen en gitt frist der de er uenige med Legemiddelverkets miljørisikovurderinger. Ved uenighet foreslår disse utvalgsmedlemmene at uenigheten løftes til de to overordnede departementene. Som nevnt over, er utvalget samstemte i at ved søknader om utsetting ved klinisk utprøving av legemidler til mennesker og utprøving av legemidler til dyr, der det ikke skal gjøres en forenklet vurdering, skal VKM alltid foreta en risikovurdering, og Miljødirektoratet skal gis anledning til å gi innspill.
I lys av at utvalget har delte meninger om vedtaksmyndighet etter GMO-regelverket, presenteres under fordeler og ulemper ved alternativene.
12.7.3.1 Legemiddelverket som vedtaksmyndighet for GMO-legemidler til både mennesker og dyr
Legemiddelverket er koordinerende etat og har mer dokumentasjon tilgjengelig for søknaden i kraft av at etaten også godkjenner den kliniske utprøvingen etter legemiddelregelverk. All dokumentasjon som kreves for en utsettingssøknad skal uansett sendes til vedtaksmyndigheten, men ettersom Legemiddelverket allerede har dokumentasjon på sikkerhet, effekt, og produksjonskvalitet, er det sannsynlig at denne dokumentasjonen i de fleste tilfeller dekker eventuelle behov for ytterligere opplysninger til miljørisikovurderingen. I disse tilfellene vil derfor Legemiddelverket ikke ha behov for å etterspørre informasjon fra søker. Dette vil bidra til raskere og mer effektiv saksbehandling. Etaten har god oversikt over regelverk som angår klinisk utprøving, både nasjonalt og i EU/EØS. Legemiddelverket som vedtaksmyndighet vil også følge det ordinære sektorprinsippet.
Legemiddelverket har erfaring med å vurdere miljørisiko i forbindelse med søknader om markedsføringstillatelse, men kun for GMO-legemidler til mennesker, og ikke til dyr. I forbindelse med overføring av vedtaksmyndighet, opprettet Legemiddelverket i årsskiftet 2021/22 samarbeid med VKM om risikovurdering av GMO-legemidler til klinisk utprøving både hos dyr og mennesker. Utvalgets ønske om at VKM skal gjøre miljørisikovurdering av GMO-legemidler som ikke kvalifiserer for forenklet vurdering, ivaretas derfor i en modell der Legemiddelverket er vedtaksmyndighet for studier både på mennesker og dyr.
12.7.3.2 Miljødirektoratet som vedtaksmyndighet for GMO-legemidler til dyr
Miljødirektoratet er ansvarlig myndighet for alle andre typer utsetting av GMO, og har god oversikt over regelverk og krav til utsetting, både nasjonalt og praksis i EU/EØS. Miljødirektoratet har siden 2017 hatt løpende oppdrag til VKM om å utføre helse- og miljørisikovurdering38, og har hatt som rutine at VKM foretar risikovurdering for alle søknader om klinisk utprøving. Det er også Miljødirektoratet, som nasjonal GMO-myndighet, som spiller inn i miljørisikohøring sentralt i EU ved søknader om markedsføringstillatelse. Her har søknader om GMO-legemidler til både mennesker og dyr blitt vurdert. Miljødirektoratet har som sådan opparbeidet seg erfaring i å håndtere miljørisiko for GMO-legemidler, også til dyr, og til å vurdere eventuelle vilkår i en tillatelse.
Med endring av vedtaksmyndighet fra 15. november 2021, skal Miljødirektoratets uttalelse innhentes som grunnlag for beslutningen, og tillegges avgjørende vekt i miljørisikovurderingen. For å ivareta sin rolle her, har Miljødirektoratet sett at direktoratet fortsatt bør benytte VKM som organ for miljørisikovurdering. Dagens saksbehandlingsrutiner legger til rette for at Miljødirektoratet kan gi oppdrag til VKM for risikovurdering av GMO-legemidler til dyr. Miljødirektoratet anser at det er spesielt viktig at VKM involveres i risikovurderingene av GMO-legemidler som skal brukes på dyr. Dersom Miljødirektoratet ser behov for ytterligere opplysninger fra søker i forbindelse med risikovurdering av klinisk utprøving av GMO-legemidler til dyr, må dette med Legemiddelverket som vedtaksmyndighet gjøres via Legemiddelverket. Bruk av mellomledd kan innebære mer omstendelig saksbehandling enn direkte kontakt mellom direktoratet og søker.
12.7.3.3 Etatenes vurdering om bruk av VKM
Utvalget anser det som hensiktsmessig at vurdering av miljørisiko og håndtering av miljørisiko er tydelig adskilt i forvaltningen, for å sikre at risiko vurderes av fageksperter, og at disse er uavhengige. Begge etater (Miljødirektoratet og Legemiddelverket) ønsker å bruke VKM som risikovurderingsorgan, slik at et slikt skille opprettholdes. Det er viktig at alle etater bidrar til åpenhet om risikovurderingen, og åpent tilgjengelige risikovurderinger er en del av dette.
Dagens saksbehandlingsrutiner mellom Miljødirektoratet og Legemiddelverket legger opp til at Legemiddelverket skal vurdere fra sak til sak om VKM skal involveres. Legemiddelverket har etter at rutinene ble ferdigstilt blitt enige med VKM om at alle søknader om miljørisikovurdering skal sendes til VKM for vurdering. Dette gjelder både legemidler til mennesker og dyr.
12.7.3.4 Vedtaksmyndighet
Flertallet mener det er viktig å samle vedtaksmyndigheten for alle kliniske studier med legemidler til både mennesker og dyr hos én etat, og at dette bør være Legemiddelverket. Dette vil sørge for enhetlig forvaltning og (ressurs)effektive godkjenningsprosesser på tvers av legemiddelområdet, i tråd med sektorprinsippet. Det er i alle tilfeller VKM som skal gjøre risikovurderingene, som forklart i foregående delkapittel. Beslutningsgrunnlaget vil derfor være det samme uavhengig av hvilken etat som er oppdragsgiver, og flertallet forventer at konklusjonene om risiko også vil bli de samme uavhengig av hvilken etat som er vedtaksmyndighet. Dersom risikovurderingene fra VKM samt hensyn til dyrehelse/-velferd (som ivaretas av generelt regelverk) ikke tilsier at det er grunn til å avvise en studie, bør dette legges til grunn og utsetting av GMO-legemidlet i studien godkjennes. Dette er i tråd med prinsippet om faglig uavhengighet og at risikovurdering og risikohåndtering holdes adskilt. Godkjenning av kliniske studier bør ikke gjøres til gjenstand for politisk beslutning.
12.7.3.5 Delt vedtaksmyndighet
For GMO-legemidler til dyr, går et mindretall av utvalget inn for en ordning som innebærer at Miljødirektoratet gis en mulighet til å stanse godkjenningen innen en gitt frist der direktoratet er uenig med Legemiddelverkets innstilling i miljørisikovurderinger. Ved uenighet foreslår disse utvalgsmedlemmene at uenigheten løftes til de to overordnede departementene. Mindretallet legger vekt på miljøhensyn, og anser at utprøving av GMO-legemidler til dyr reiser særlige problemstillinger knyttet både til bærekraft og miljø. Miljødirektoratet som fagorgan på miljøområdet anses som mest egnet til å håndtere disse utfordringene. Basert på argumentene presentert over, samt Miljødirektoratets langvarige erfaring med vurdering av bærekraft, samfunnsnytte og etikk, vurderer dette mindretallet det som en hensiktsmessig løsning at Legemiddelverket er vedtaksmyndighet for klinisk utprøving av GMO-legemidler til mennesker, men for klinisk utprøving av GMO-legemidler til dyr, vil det være delt vedtaksmyndighet som beskrevet over.
12.7.4 Utvalgets vurderinger av foreslått modell
Slik modellen nå legger opp til, vil det sammenlignet med dagens praksis være flere studier som kan få godkjenning som forenklet utsetting. Forenklet utsetting er kun foreslått å gjelde bestemte GMO-legemidler til mennesker, og ikke til dyr. Dette fordi søknadsskjemaene som er foreslått brukt for å angi hvorvidt et GMO-legemiddel har neglisjerbar risiko eller ikke, kun er utarbeidet for GMO-legemidler til mennesker. Dersom det i fremtiden utvikles tilsvarende søknadsskjemaer for GMO-legemidler til dyr, vil det også kunne føre til klassifisering som forenklet utsetting. Ved saksbehandling som forenklet utsetting av GMO-legemidler til mennesker har ikke Miljødirektoratet noen formell rolle, og inkludering av færre etater vil effektivisere saksbehandlingen. Slike studier krever kun melding når det gjelder GMO-aspektet, og derfor vil det antakelig heller ikke være krav om offentlig høring.
Utvalget anser at innføring av foreslått modell for søknadsbehandling av klinisk utprøving av GMO-legemidler (forenklet utsetting), i hovedsak vil ha positive virkninger. Ingen negative virkninger av betydning er identifisert. Eventuelle behov for endringer i foreslått modell, vurderer utvalget at relativt enkelt kan gjøres ved å endre forvaltningspraksis. Med unntak av å forskriftsfeste forenklet utsetting og krav til dette, er det ikke vurdert at foreslått modell krever andre regelverksendringer. Som nevnt skal vedtaksmyndighet for klinisk utprøving av GMO-legemidler både hos mennesker og dyr opp til vurdering etter at denne utredningen er lagt fram. Ved å inkludere konseptet forenklet utsetting, og krav til dette, anser utvalget at foreslått modell for godkjenning av søknader med GMO-legemidler i klinisk utprøving etterkommer søkers ønske om effektiv saksbehandling og genteknologilovens krav om vurdering av helse- og miljørisiko, på et nivå tilpasset GMO-ens risiko. Detaljer for saksbehandling i foreslått modell beskrives under i 12.7.5.
12.7.5 Detaljer for saksbehandling i foreslått modell
I foreslått modell er Legemiddelverket inngangsport for søkere, samt at Legemiddelverket koordinerer saksbehandlingen mellom involverte etater; Legemiddelverket, Helsedirektoratet og Miljødirektoratet. Søker sender både melding og søknad til samme mottaker, Legemiddelverket.
Legemiddelverket samkjører frister for saksbehandling etter genteknologiloven tilpasset fristene satt i sentral prosedyre i forordning (EU) nr. 536/2014 for kliniske studier, og veterinærforordning (EU) nr. 2019/6. Det følger av forordning (EU) nr. 536/2014 at studien skal vurderes av en etikkomite. Den kliniske utprøvingen må godkjennes etter legemiddelregelverkene i tillegg til godkjenning etter genteknologiloven.
I tabell 12.1 er det skissert alternative vedtaksmyndigheter, enten Legemiddelverket (a), eller delt vedtaksmyndighet b), der det foreslås at Miljødirektoratet gis en mulighet til å stanse godkjenningen innen en gitt frist der direktoratet er uenig med Legemiddelverkets innstilling i miljørisikovurderingen. Legemiddelverket er vedtaksmyndighet for GMO-legemidler til mennesker.
Tabell 12.1 Oversikt over alternativer a) og b) i foreslått modell
Modell navn Min – maks tid | Hvilken etat møter søkerne først | Myndighet som vurderer melding (er krav til forenklet utsetting oppfylt?) | Rolle Hdir | Hvem fatter vedtak ved utsetting av GMO-legemidler? | VKMs risikovurdering ved utsetting | BSE-vurdering ved utsetting? | Alle søknads skjema benyttes |
|---|---|---|---|---|---|---|---|
55-105 D | SLV | SLV, innen dag 5a | Hdir godkjenner ved kravb for håndtering under innesluttet bruk | fra dag 2 – dag 32, minimum 30 dager c | kun GMO-legemidler til dyr | Ja | |
a) | SLV for mennesker og dyr | ||||||
b) | SLV for mennesker, både SLV og Mdir for dyr |
Tidene i tabellen er kalenderdager, og er basert på tidene til forordning (EU) 536/2014 for kliniske studier, og forordningen om legemidler til dyr (EU) 2019/6 (validering + 45D vurdering). Vurdering av REK-KULMU er en del av godkjenningen etter legemiddelregelverk. Vurdering av GMO-aspekter etter direktiv 2001/18/EF er ikke tatt inn i forordningen, men det vurderes hensiktsmessig å samkjøre saksbehandlingstider for å få en best mulig prosess i Norge. Dersom legemidlet er avansert terapi (ATMP), som kan være GMO, kan tidene forlenges (55 dager (D) + 50D). Rapportørlandet, som er ansvarlig for søknadsbehandlingen, avgjør dette. Alle modellene vil ha like frister for saksbehandling. Hdir – Helsedirektoratet, Mdir – Miljødirektoratet, SLV – Legemiddelverket, REK-KULMU, VKM – Vitenskapskomitéen for mat og miljø. BSE-vurdering – vurdering av bærekraft, samfunnsnytte og etikk etter genteknologiloven
a. Tilpasset valideringstiden.
b. Krav til håndtering under innesluttet bruk-reglene slår inn dersom GMO-en er replikasjonskompetent og legemidlet ikke leveres til ferdig bruk, men må tilberedes før administrasjon til pasient. Begge kriterier må være oppfylt.
c. Tiden til VKMs risikovurdering er tilpasset saksbehandlingstiden på 45 dager, eventuelt med forlengelse (+50 dager) grunnet innhold av GMO i legemidler til mennesker. Den nye veterinærforordningen inneholder ingen bestemmelse for slik forlengelse.
12.7.5.1 Veivalg 1: Melding – forenklet utsetting
For søknader om klinisk utprøving med GMO-legemidler som oppfyller krav til forenklet utsetting, kreves kun melding. Alle meldinger inkludert dokumentasjon videresendes til orientering til Helsedirektoratet og Miljødirektoratet automatisk. Myndigheten som vurderer denne meldingen, er Legemiddelverket, og dette gjøres innen fem arbeidsdager. Legemiddelverket kan vurdere å konferere med Helsedirektoratet og Miljødirektoratet om denne vurderingen dersom Legemiddelverket ser behov for det. Helsedirektoratet vurderer om studien krever godkjenning etter annet helseregelverk, for eksempel forskrift om håndtering av humane celler og vev. Som nevnt over svarer Legemiddelverket søker innen 30 kalenderdager, med bekreftelse på at forenklet utsetting er dokumentert, og at ingen tillatelse til utsetting etter genteknologiloven er påkrevd. Søker kan ikke starte utprøvingen før Legemiddelverket har svart. I de tilfeller Legemiddelverket ikke svarer innen fristen, skal dette tolkes som at søker kan gå i gang med den kliniske studien hva gjelder tillatelser til utsetting etter genteknologiregelverket. Legemiddelverket kan ved behov sette eventuelle risikohåndteringskrav i sitt svar til søker. I tillegg skal lokalene utprøvingen foregår i godkjennes av Helsedirektoratet der det er relevant.
Kliniske studier der utprøvingslegemidlet består av, eller inneholder humane genmodifiserte celler, eller adeno-assosiert virus reguleres i foreslått modell også som forenklet utsetting, der krav om at GMO-legemidlet kan anses å føre til biologisk inneslutning er tilfredsstilt. Her kreves kun melding.
Gangen videre i de tilfeller Legemiddelverket vurderer at krav til forenklet utsetting ikke er oppfylt, er som følger: Legemiddelverket varsler søker, Miljødirektoratet og Helsedirektoratet innen dag 5 (se tabell 12.1). Miljødirektoratet og Helsedirektoratet har på dette tidspunktet allerede mottatt all dokumentasjon fra søker. Se videre om gangen frem mot vedtak for utsettingssøknader i 12.7.5.2 under.
Utvalget er samstemte i anbefalingen om en slik ordning/kategori, og at EU-skjemaene skal være førende for hvilke typer GMO-legemidler som kvalifiserer for forenklet utsetting.
12.7.5.2 Veivalg 2: Søknad om utsetting
Legemiddelverket videresender, som for melding beskrevet i 12.6.4.2 over, automatisk all dokumentasjon til Miljødirektoratet og Helsedirektoratet, senest innen to arbeidsdager. Legemiddelverket angir frister tilpasset frister i søknadsprosedyrene for klinisk utprøving (mennesker og dyr). Legemiddelverket vurderer om søknaden eventuelt kan kreve tillatelse til innesluttet bruk, og involverer i så fall Helsedirektoratet. Vedtak etter genteknologiloven skal fattes innen dag 55 (se figur 12.1 og tabell 12.1).
Alternativ a) Legemiddelverket er vedtaksmyndighet
Legemiddelverket vurderer om søknaden om utsetting er valid, og Legemiddelverket fatter vedtak om utsetting etter genteknologiloven. Det innhentes uttalelse fra Miljødirektoratet som grunnlag for beslutningen, og i miljørisikovurderingen skal det legges avgjørende vekt på uttalelsene fra miljømyndighetene. Legemiddelverket gir oppdrag til VKM om å foreta miljørisikovurdering for søknader om klinisk utprøving til mennesker og dyr. Tillatelsen gis med eventuelle vilkår avhengig av risikovurderingen.
Alternativ b) Delt vedtaksmyndighet
For søknader om klinisk utprøving av GMO-legemidler til mennesker, likt som over i a). For søknader om klinisk utprøving av GMO-legemidler til dyr, er det en ordning som innebærer at Miljødirektoratet gis en mulighet til stanse godkjenningen innen en gitt frist der de er uenige med Legemiddelverkets miljørisikovurderinger. Ved uenighet foreslås det at uenigheten løftes til de to overordnede departementene.
Et flertall på 7 (Arne Holst-Jensen, Espen Gamlund, Muath Alsheikh, Trygve Brautaset, Camilla Tøndel, Anna Wargelius, Sigrid Bratlie) i utvalget anbefaler alternativ a) der Legemiddelverket beholder vedtaksmyndigheten alene i tråd med gjeldende regler som trådte i kraft etter overføringen av vedtaksmyndighet i november 2021. Et mindretall på 4 medlemmer (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) ønsker delt vedtaksmyndighet som skissert i b).
12.7.5.3 Veivalg 3: Søknad om utsetting for GMO som krever håndtering under innesluttet bruk
Noen GMO-legemidler vil kreve tillatelse etter både innesluttet bruk- og utsettingsregelverket. For eksempel dersom GMO-en som omsøkes i studien inneholder en replikasjonskompetent virusvektor, og legemidlet ikke leveres til ferdig bruk, men må tilberedes før det gis til pasient. Begge kriterier må være oppfylt for at kravet skal komme til anvendelse. Det kan være hensiktsmessig at også dette kravet spesifiseres i forskrift, og det kan gjøres samtidig som krav til biologisk inneslutning spesifiseres i forskrift.
Som for veivalg 2, videresender Legemiddelverket automatisk all dokumentasjon til Miljødirektoratet og Helsedirektoratet, senest innen to arbeidsdager. Legemiddelverket angir frister tilpasset frister i legemiddelregelverket.
Helsedirektoratet vurderer hvilket inneslutningsnivå som vil kreves, med utgangspunkt i dokumentasjonen fra søker. Dersom fasiliteten som skal brukes ikke allerede har godkjenning, vurderer Helsedirektoratet søknaden innen dag 45 for inneslutningsnivå 1 og 2 (tilsvarer dagens frist i forskrift for innesluttet bruk av GMO).
For beskrivelse av videre saksgang og vedtaksmyndighet, se 12.7.5.2.
Utvalget stiller seg samlet bak dette forslaget. I figur 12.3 vises en skjematisk oversikt over forslag til saksgangen for søknader og saksbehandling for kliniske utprøvinger med GMO-legemidler. Den første viser for utprøving til mennesker og den andre for utprøving til dyr.
12.7.5.4 Søknadsskjema
Interplay-samarbeidet i EU, hvor formålet er å harmonisere praksis i de ulike EU-landene hva gjelder GMO-godkjenningene for kliniske studier med GMO-legemidler, har vært viktig for operasjonaliseringen og praksisen i Norge under GMO-regelverket.
Det er utviklet veiledninger og søknadsskjema, som er frivillige å ta i bruk i hvert enkelt land, og søknadskjemaene kan brukes etter regelverk om både innesluttet bruk og utsetting. Det angis hvilke land søknadsskjemaene kan benyttes i. Veiledningene og skjemaene oppfattes som forenklende både for søker og myndigheter. De aller fleste medlemslandene har gitt sin tilslutning til alle GMO-interplay dokumentene, i likhet med Norge.
Med avklaringene gjort mellom Helsedirektoratet og Miljødirektoratet i 2018, angående at utsetting gjelder fra GMO-en er satt inn i/administrert til testpersonene og disse beveger seg ut fra de innesluttede fasilitetene, har søknadsskjemaer først og fremst vært tenkt brukt til å søke godkjenning som utsetting.
Foreslått modell legger opp til at alle søknadskjema kan brukes. Avhengig av GMO-legemiddel benyttes disse til å behandle søknader etter regelverk om innesluttet bruk eller utsetting. Bruk av søknadsskjema harmonerer med praksis i EU/EØS, og det er forenklende for både søker og myndigheter.
12.7.5.5 Vurdering av bærekraft, samfunnsnytte og etikk (BSE)
I foreslått modell legges det opp til at søknader om utsetting av GMO i legemidler benyttet i klinisk utprøving til mennesker ikke skal vurderes etter genteknologilovens kriterier om bærekraft, samfunnsnytte og etikk. Utvalget anser at vurderingen er tilstrekkelig dekket i legemiddelregelverk, gjennom vurdering av SLV og REK KULMU (Regional komite for klinisk utprøving av legemidler til mennesker).
Flertallet (Arne Holst-Jensen, Espen Gamlund, Muath Alsheikh, Trygve Brautaset, Camilla Tøndel, Anna Wargelius, Sigrid Bratlie) mener at det heller ikke skal foretas vurdering av BSE ved klinisk utprøving av legemidler til dyr. I kliniske studier på dyr mener flertallet at hensyn knyttet til dyrevelferd ivaretas av annet regelverk og vil alltid være en del av godkjenningen for en klinisk studie. Ellers viser flertallet til samme argumentasjon som et samlet utvalg vektlegger i sin felles anbefaling om å ikke kreve vurderinger av etisk forsvarlighet ved forsøksutsetting av GMO/PB dyr, planter og mikroorganismer til andre formål (kapittel 10): På forsøksstadiet er det ikke nødvendigvis generert slike data, og selve poenget med en forsøksutsetting er å samle data og opparbeide dokumentasjon som senere skal brukes i en søknad om godkjenning.
Mindretallet (Fern Wickson, Ingvild Ulrikke Jakobsen, Kåre Magne Nielsen og Aina Bartmann) mener det fortsatt skal foretas en vurdering av etisk forsvarlighet, inkludert bærekraft, når det gjelder GMO-legemidler til dyr. Mindretallet antar at mange søknader vil være knyttet til å øke effektiviteten til produksjonsdyr, og kan reise særlige problemstillinger. Videre vil omfang og tempo kunne sammenlignes med utsetting fordi det dreier seg om store populasjoner og et globalt marked med stor spredning. Det er derfor viktig å kunne vurdere etisk forsvarlighet etter genteknologiloven helt fra starten av. Mindretallet viser også til at da Klima- og miljødepartementet sendte på høring forslag om en slik endring i genteknologiloven i 2020, var det en rekke høringsinstanser som gikk mot at dette skulle gjelde for dyr, fordi de anså at annet regelverk ikke ivaretok BSE-kriteriene godt nok. Det er per i dag ingen klar operasjonalisering av vurdering av BSE-kriterier for GMO-legemidler, hverken for mennesker eller dyr. Mindretallet vurderer at en egnet instans bør utarbeide tilpassede retningslinjer for denne vurderingen.
12.7.5.6 Høring
Mange søknader vil med foreslått modell behandles som forenklet utsetting. Flertallet mener at dette ikke skal kreve høring. For søknader om kliniske studier med GMO-legemidler til utsetting som foreslås å være godkjenningspliktige, anbefaler flertallet på 7 medlemmer (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) at det heller ikke gjennomføres høring, verken når det gjelder legemidler til mennesker eller dyr. Dette er i tråd med flertallets anbefaling om at forsøksutsetting av PB- og GMO-organismer til annen bruk ikke bør høres, som beskrevet i kapittel 10.2. Kliniske studier er også forsøksutsetting.
Flertallet mener at det er tilstrekkelig å legge ut informasjon om søknaden på nettsidene til ansvarlig forvaltningsinstans (Legemiddelverket), i tråd med praksis i Sverige og Tyskland. Videre viser dette flertallet til at det som regel ikke kommer noen høringssvar på saker om GMO-legemidler, og anser at hensyn knyttet til åpenhet og informasjon til offentligheten derfor ivaretas tilstrekkelig gjennom foreslåtte informasjonsordning. Flertallet mener at det ikke er noe prinsipielt problematisk ved GMO-legemidler sammenlignet med andre legemidler, og at relevante hensyn som bør ligge til grunn for vurdering av søknader om kliniske studier er av faglig karakter. Dette mener flertallet best ivaretas av kompetente myndigheter og ikke offentligheten for øvrig. Flertallet viser videre til at en slik endring forenkler saksbehandlingen og korter ned tiden det tar å få en godkjenning etter genteknologiregelverket på plass. Dette vil både frigjøre ressurser i forvaltningen som kan brukes på andre oppgaver samt bidra til å gjøre Norge mer attraktivt for kliniske studier med genterapi og andre GMO-legemidler fordi prosessen vil være mindre krevende for søker.
Forslaget om å unnlate høring for GMO-legemidler, både til forenklet utsetting (melding) og godkjenningspliktige søknader om utsetting, vil sannsynligvis kreve lovendring.
Et mindretall på 4 medlemmer (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) går inn for et fortsatt krav om å ha høring for søknader om utsetting som angår klinisk utprøving av GMO-legemidler til mennesker (både meldinger om forenklet utsetting og øvrige søknader om utsetting). Dette mindretallet anser at et fortsatt krav om høring for søknader om utsetting bidrar til åpenhet, tillit og saksopplysning, og at høring enkelt gjennomføres digitalt innenfor avsatt saksbehandlingstid. Dette er i tråd med formålet til åpenhetsforordningen som nylig har trådt i kraft i EU, og prinsippet om mer offentlighet i norsk forvaltning. Basert på argumentene over, vurderer dette mindretallet av utvalget at nytten av en høring for utsettingssøknader veier tyngre enn at søker eventuelt kan oppleve høring som belastende
Et mindretall på fire utvalgsmedlemmer (Aina Bartmann, Fern Wickson, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen) stiller seg bak forslaget om at det skal gjennomføres høring ved klinisk utprøving av GMO-legemidler til dyr ved søknader om utsetting av GMO etter genteknologiloven. Begrunnelsen er den samme som for klinisk utprøving av legemidler til mennesker som er beskrevet over.
12.8 Oppsummering av anbefalingene
Forenklet vurdering – samlet anbefaling (se nærmere begrunnelse i 12.6.4)
Et samlet utvalg anbefaler at alle EUs ferdigutviklede søknadsskjemaer kan benyttes, og alle GMO-legemidler som kvalifiserer for skjema kan ha en forenklet behandling hos vedtaksmyndigheten. I praksis innebærer dette en melding til myndigheten. Foreløpig er disse skjemaene bare utviklet for vurdering av GMO-legemidler til mennesker.
Regelverket – delt anbefaling (se nærmere begrunnelse i 12.6.2)
Flertallet (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) mener at GMO-legemidler i sin helhet bør reguleres i legemiddelregelverket og forvaltes av helsemyndighetene.
Mindretallet (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) mener at GMO-legemidler fremdeles bør reguleres under genteknologiloven.
Vedtaksmyndighet – delt anbefaling (se nærmere begrunnelse i 12.6.2)
Utvalget er samlet om at Legemiddelverket skal være inngangsport for søknaden
Utvalget er også samlet om at Legemiddelverket skal være vedtaksmyndighet for klinisk utprøving av GMO-legemidler til mennesker.
Flertallet (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) anbefaler at Legemiddelverket har vedtaksmyndighet også for vurdering av søknader om bruk av GMO-legemidler i klinisk utprøving til dyr.
Mindretallet (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) anbefaler delt vedtaksmyndighet for klinisk utprøving av GMO-legemidler til dyr. Det vil si at Miljødirektoratet gis en mulighet til å stanse godkjenningen innen en gitt frist der direktoratete er uenig med Legemiddelverkets innstilling i miljørisikovurderinger. Ved uenighet foreslår disse utvalgsmedlemmene at uenigheten løftes til de to overordnede departementene.
Høringer – delt anbefaling (se nærmere begrunnelse i 12.7.5.6)
Mange søknader vil med foreslått modell behandles som forenklet utsetting. Flertall mener at dette ikke skal kreve høring. For søknader om kliniske studier med GMO-legemidler til utsetting som foreslås å være godkjenningspliktige, anbefaler flertallet på 7 medlemmer (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) at det heller ikke gjennomføres høring, verken når det gjelder legemidler til mennesker eller dyr.
Et mindretall på 4 medlemmer (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) går inn for et fortsatt krav om å ha høring for søknader om utsetting som angår klinisk utprøving av GMO-legemidler til mennesker (både meldinger om forenklet utsetting og øvrige søknader om utsetting). Det samme mindretallet anbefaler også høring ved klinisk utprøving av GMO-legemidler til dyr.
Vurdering av bærekraft, sambunsnytte og etikk (BSE) – delt anbefaling (Se nærmere begrunnelse i 12.6.6 og 12.7.5.5)
Utvalget er samlet i at det ikke skal være BSE-vurdering for GMO-legemidler i klinisk utprøving til mennesker. Ved kliniske studier på mennesker ivaretas etikkvurderinger av REK KULMU, uavhengig av typen legemiddel.
Flertallet (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) mener at vurdering av BSE ikke skal gjelde for GMO-legemidler i klinisk utprøving til dyr. Flertallet mener at hensyn knyttet til dyrevelferd ivaretas av annet regelverk og vil alltid være en del av godkjenningen for en klinisk studie. Ellers viser flertallet til samme argumentasjon som et samlet utvalg vektlegger i sin felles anbefaling om å ikke kreve vurderinger av etisk forsvarlighet ved forsøksutsetting av GMO/PB dyr, planter og mikroorganismer til andre formål (kapittel 10): På forsøksstadiet er det ikke nødvendigvis generert slike data, og selve poenget med en forsøksutsetting er å samle data og opparbeide dokumentasjon som senere skal brukes i en søknad om godkjenning.
Mindretallet (Aina Bartmann, Ingvild Ulrikke Jakobsen, Kaare Magne Nielsen og Fern Wickson) mener det fortsatt skal foretas en vurdering av BSE, når det gjelder GMO-legemidler til dyr. Mindretallet antar at mange søknader vil være knyttet til å øke effektiviteten til produksjonsdyr, og kan reise særlige problemstillinger. Videre vil omfang og tempo kunne sammenlignes med utsetting fordi det dreier seg om store populasjoner og et globalt marked med stor spredning. Det er derfor viktig å kunne vurdere etisk forsvarlighet etter genteknologiloven helt fra starten av. Det er per i dag ingen klar operasjonalisering av vurdering av BSE-kriterier for GMO-legemidler, hverken for mennesker eller dyr. Mindretallet vurderer at en egnet instans bør utarbeide tilpassede retningslinjer for denne vurderingen
Innesluttet bruk (Se nærmere begrunnelse i 12.6.3)
Flertallet (Anna Wargelius, Muath Alsheikh, Sigrid Bratlie, Trygve Brautaset, Espen Gamlund, Arne Holst-Jensen og Camilla Tøndel) anbefaler at regler for innesluttet bruk av GMO gjennomgås i en egen prosess i oppfølgingen av denne NOU-en.
Fotnoter
https://www.dagensmedisin.no/lov-og-rett/kreftstudie-satt-pa-vent-absurd-situasjon/208810
https://eur-lex.europa.eu/legal-content/DA/TXT/?uri=celex%3A32001L0018
Genteknologiloven
Accelerating Clinical Trials in the EU
Nasjonal handlingsplan for kliniske studier 2021-2025
EU-Kommisjonens strategi om medisiner i miljøet (2019): https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52019DC0128&from=EN
EU-Kommisjonens forslag til ny legemiddelstrategi https://health.ec.europa.eu/medicinal-products/pharmaceutical-strategy-europe_en
Klima- og miljødepartementet, høring av forslag til endring av genteknologiloven: https://www.regjeringen.no/no/dokumenter/horing---forslag-til-endringer-i-genteknologiloven/id2701165/?expand=horingssvar
Norge har kun deltatt aktivt i gruppe 1, gruppen kalt GMO-pharma. Norge har ikke deltatt i interplay-samarbeidet om veterinære GMO-legemidler.
Forskrift om krav til kvalitet og sikkerhet ved håndtering av humane celler og vev
Lov 5.desember 2003 om humanmedisinsk bruk av bioteknologi m.m nr.100
En replikasjonskompetent virusvektor kan fortsatt formere seg (replikere), den inneholder altså alle DNA-elementer som trengs for å kunne replikere.
Resterende virusvektor-partikler er kun deler av den opprinnelige virusvektoren, og ikke en fullstendig virusvektor. Slike partikler kan ikke replikere.
Søknadsskjemaet har blitt revidert til å inkludere flere bruksområder. Siste revisjon var i september 2021.
https://ec.europa.eu/health/medicinal-products/advanced-therapies_en
Alle VKMs vurderinger og mer info om oppdraget kan finnes på VKMs nettsider VKM har vurdert EUs harmoniserte søknadsskjemaer for klinisk utprøving av GMO-legemidler til mennesker – Vitenskapskomiteen for mat og miljø
Følgende må være oppfylt for at miljørisiko skal kunne vurderes som neglisjerbar: (1) ingen skadelige DNA-elementer er tilstede i vektoren, (2) det kan dokumenteres at det ferdige legemidlet ikke inneholder replikerende virusvektorer, og nivået av eventuelle resterende virusvektor-partikler er på et neglisjerbart nivå, og (3) spesifikt for humane genmodifiserte celler; søknadsskjemaet gjelder kun for celler modifisert med bestemte virusvektorer.
COGEMs vurdering datert 20.08.2021 https://cogem.net/app/uploads/2021/09/210820-01-Advice-EU-consultation-GMOs-in-medical-applications.pdf
Impact of genetically modified organism requirements on gene therapy development in the EU, Japan, and the US: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9207611/
Nasjonale skjema om godkjenning av klinisk utprøving av GMO-legemidler til mennesker: Genetically Modified Organism (GMO) aspects for investigational medicinal products Public Health (europa.eu)
https://ec.europa.eu/health/system/files/2017-04/vol6c_gmo_guidance_2017_03_0.pdf
https://legemiddelverket.no/veterinærmedisin/klinisk-utproving-av-legemidler-til-dyr
Lenke til instruksen
For mer informasjon om komitéens arbeid, se Forsøksdyrkomitéen (forsoksdyrkomiteen.no)
Lenke til retningslinjen
Eksempler på slike veterinære legemidler er vekstfremmende hormoner, (inkludert for økt melkeproduksjon), antibiotika som vekstfremmer eller som forebygging mot sykdom, antibiotika viktige for human bruk, og legemidler til annen bruk enn det er godkjent for (off-label), samt for andre hensyn enn dyrehelse og dyrevelferd.
Legemiddelverket fatter vedtak etter legemiddelregelverk, som inkluderer behandling av REK-KULMU. Legemiddelverket fatter også, som midlertidig ordning, vedtak etter genteknologiloven for tillatelse til utsetting av GMO. Dersom studien skal utføres i lokaler som ikke har godkjenning, må dette søkes om til Helsedirektoratet.
Kommisjonens hjemmeside om ny strategi: https://health.ec.europa.eu/medicinal-products/pharmaceutical-strategy-europe_en
Begrepet tilberedning er definert i legemiddelhåndteringsforskriften § 3 bokstav j) som enkel tilvirkning av legemiddel som på grunn av kort holdbarhet, må gjøres bruksferdig umiddelbart før bruk. Enkel istandgjøring er det som tilberedes rett før utlevering til pasient, f.eks på grunn av kort holdbarhet, og er omtalt i den samme forskriften, i § 4. https://lovdata.no/dokument/SF/forskrift/2001-06-26-738
En oversikt over forslag til risikohåndteringstiltak kan finnes i følgende dokument utarbeidet i GMO-interplay, i forbindelse med generell bruk av onkolytiske virus: Considerations for the evaluation of shedding (2020), side 5-6, chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://health.ec.europa.eu/system/files/2022-01/oncolytic_evaluation_en.pdf
https://www.hse.gov.uk/biosafety/gmo/acgm/acgmcomp/part6.pdf
Hvorvidt de humane genmodifiserte cellene ikke lenger omfattes av genteknologiloven etter at de er satt tilbake i testpersonen, er en gråsone i lovverket. Praksis har vært å forstå loven som at cellene ikke lenger er omfattet etter innsettelse. En genmodifisert virusvektor vil alltid være omfattet av loven, uavhengig av om den befinner seg i celler i kultur eller i et menneske.
Dette fraværet dokumenteres på produksjonsnivå, måten GMO-en framstilles på skal sikre at replikerende virusvektorer ikke er tilstede i det ferdige GMO-legemidlet.
VKM har i sin vurdering av dette søknadsskjemaet omtalt at miljørisiko vurderes for AAV-vektorer til å være lav/neglisjerbar. Sentralt for denne vurderingen er at AAV-vektorer formerer seg i en celle kun når det foreligger en trippel infeksjon av cellen.
Som tidligere nevnt, klinisk utprøving er ikke definert som forskning i forsøksdyrforskriften, og derav kommer ikke denne forskriften til anvendelse ved klinisk utprøving. Men for praktisk forståelse av klinisk utprøving kan dette sees på som forskning, og formålet er å innhente data til senere bruk i søknader om markedsføringstillatelse for legemidlet.
Deliberate release into the environment of other than plants GMOs for any other purposes than placing on the market (experimental releases): https://gmoinfo.jrc.ec.europa.eu/gmo_browse.aspx
Søknader etter legemiddelregelverket og GMO-regelverket sendes samlet til legemiddelmyndigheten, og legemiddelmyndigheten fatter vedtak om utsettingssøknaden.
Genteknologiloven har krav om å vurdere både helse- og miljørisiko ved søknad om utsetting av GMO, slik en klinisk utprøving ofte innebærer. Miljødirektoratets oppdrag til VKM inkluderte derfor både en helse- og miljørisikovurdering da direktoratet var vedtaksmyndighet etter genteknologiloven.