Del 2
Rammevilkår, utfordringer og tiltak på legemiddelområdet
3 Legemiddelpolitiske mål
3.1 Mål for legemiddelpolitikken
Legemiddelpolitikken er karakterisert ved et sterkt offentlig og statlig engasjement både når det gjelder tiltak som skal sikre legemidlenes kvalitet, sikkerhet og effekt, og ved at tilgjengelighet til legemidler skal være mest mulig uavhengig av individenes betalingsevne og bosted. I St meld nr 41 for 1987-88, «Helsepolitikken mot år 2000», er de helsepolitiske målene for apotekvesenet formulert. Det heter blant annet at:
«Hovedmålet for apotekvesenet må være å yte samfunnet best mulig farmasøytisk service. Dette krever at apotektjenesten ses i sammenheng, planlegges og utbygges i takt med det øvrige helsestell. (...) Innenfor den overordnede målsetting må apotekvesenet bidra til:
at befolkningen i alle deler av landet får rimelig lett og sikker adgang til effektive og høyverdige legemidler til lavest mulig pris og med ens utsalgspris over hele landet.
at ressursene blir hensiktsmessig fordelt innenfor legemiddelsektoren. Videre bør apotekvesenet medvirke til betryggende forhold på felter som grenser opp med legemiddelområdet».
Med særlig henblikk på apotekfunksjonen ble målsettingen konkretisert i følgende forhold:
«Det må sikres tilstrekkelig tilgang på legemidler. Befolkningen i alle deler av landet må ha rimelig lett adkomst til apotektjeneste. Publikum bør få den service de har behov for uten unødig forsinkelse.
Legemidlene må være av god kvalitet. Sikkerhetstiltakene i systemet og apotekenes ekspedisjonsrutiner må være betryggende.
Det må treffes tiltak som kan sikre forsvarlig og medisinsk riktig legemiddelbruk såvel i som utenfor institusjon. Nødvendig informasjon om legemiddelspørsmål må nå frem til alt helsepersonell som har tilknytning til medisinforsyningen. Apotekene må engasjeres i en med den øvrige helsetjeneste samordnet virksomhet som har til hensikt å fremme korrekt legemiddelhåndtering og bruk i befolkningen. Informasjonsformidling og veiledningsoppgaver må bli viktige sider ved apotekfarmasøytenes arbeids- og ansvarsområde.
Apotekene bør, i den grad det er nødvendig og forholdene ligger til rette for det, yte service også på felter innenfor helseområdet hvor apotekfunksjon ikke har monopolkarakter, blant annet når det gjelder vareområder som sykepleieartikler, forbindingssaker, handikaphjelpemidler o.l. og på felter som alminnelig helseopplysningsvirksomhet, analyse- og laboratorietjenester, førstehjelp m.v.
Prisene på apotekenes ytelser må være ens over hele landet og så lave som mulig under hensyntagen til at et tilfredsstillende kvalitetsnivå skal opprettholdes. Ressursfordelingen mellom de ulike ledd innen legemiddelområdet må være rimelig i forhold til de oppgaver som skal skjøttes».
Målet om at legemiddelprisene skal være like over hele landet ble forlatt i forbindelse med revidert nasjonalbudsjett i 1994. Endringen hadde blant annet bakgrunn i et ønske om at sykehusene skulle få større frihet til å inngå gunstige prisavtaler med leverandørene gjennom bruk av anbud m.m. Det er også bestemt at dersom apotekene oppnår gunstige innkjøpsbetingelser, skal minst halvparten av rabatten tilfalle kunden. Selv om målsettingen om like priser ble forlatt, har det hele tiden, også etter 1994, vært en ordning med regulering av offentlige maksimalpriser for legemidler. Maksimalpriser for legemidler settes på grunnlag av blant annet legemiddelprisene i andre EU/EØS-land, herunder land med fri prissetting på legemidler. Legemiddelprodusentene ønsker i liten grad å selge legemidler rimelig i enkeltland, fordi det kan føre til lavere priser i andre markeder på grunn av såkalt parallelleksport. Dette gjør det vanskelig for norske myndigheter å oppnå lavere legemiddelpriser enn andre europeiske land. Siden 1995 har det vært fri prisdannelse på reseptfrie legemidler. Dette har ført til at legemiddelprodusentene har økt prisene, noe som blant annet kan skyldes manglende konkurranse i apotekmarkedet.
I ovennevnte målsettinger for apotekvesenet er målet om tilgjengelighet først og fremst formulert som et mål om geografisk tilgjengelighet. Målet om tilgjengelighet kan imidlertid også formuleres som et krav til at legemidler skal være økonomisk tilgjengelige for folk flest. Refusjonsordningene for legemidler og ordningen med egenandelstak er offentlige virkemidler for å gjøre viktige legemidler økonomisk tilgjengelige for befolkningen.
Målsettingene ble i sin tid utarbeidet for å veilede apotekpolitikken. Formuleringene har imidlertid i høy grad også fungert som generelle målsettinger for norsk legemiddelpolitikk. De omfatter blant annet krav om tilgjengelighet, faglig forsvarlig bruk og håndtering av legemidler, kvalitet og lavest mulig pris. De tre grunnleggende kravene til legemidler som medisinsk-teknologisk produkt er: kvalitet, sikkerhet og effekt.
3.2 Mål for refusjonspolitikken
Utgifter til legemidler dekkes etter forskrift fastsatt av Sosial- og helsedepartementet med hjemmel i folketrygdloven. Stønadsplikten er som hovedregel begrenset til å gjelde utgifter til medisiner til bruk ved nærmere angitte sykdommer, som er oppført i forskriften. Det generelle vilkår for refusjon er at sykdommen har gått inn i en langvarig fase, det vil si tre måneders varighet eller mer i løpet av ett år.
Grundutvalget foreslo egne mål for refusjonspolitikken for legemidler. Utvalget mente at det burde gis prioritet til legemidler til bruk mot alvorlige sykdommer, der legemidler har god effekt og er kostnadseffektive. Tilbudet må dimensjoneres til fordel for behandling der intensjonen primært er bekjempelse eller lindring av sykdom, eller forebygging hos høyrisikoindivider. På denne bakgrunn mente utvalget at refusjonsordningen for legemidler skulle bidra til å nå følgende mål:
Refusjonsordningen skal sikre mest mulig lik og enkel tilgang til effektive legemidler for små og store pasientgrupper med dokumenterte behov for medikamentell behandling.
Den skal gi samfunnet verdier for pengene, det vil si at fellesskapet skal refundere utgifter til legemidler som gir sikker helsegevinst for pasienten, og har god effekt i forhold til kostnadene. Det er viktig at kostnadsstyringen er god slik at en sikrer best mulig utnyttelse av ressursene.
Refusjonsordningen skal stimulere til ansvarlig og kostnadsbevisst forskrivning og bruk av legemidler hos lege og pasient.
Den skal medvirke til at individer med moderat og lav risiko for fremtidig sykdom, og hvor behandlingseffekten på individnivå er liten og/eller usikker, tar et størst mulig eget ansvar for egen helse. Det offentlige har først og fremst forpliktelser overfor pasienter med alvorlig sykdom eller høy risiko for sykdom, hvor det finnes effektive legemidler.
Så langt mulig skal refusjonsordningen reflektere kunnskaper om gevinster og kostnader ved bruk av legemidler, basert på helseøkonomiske undersøkelser.
Refusjonsordningen skal være enkel å administrere og forstå for legemiddelpolitikkens ulike aktører.
Ordningen skal gi myndighetene anledning til å fjerne medikamenter hvor det dokumenteres at nytten ikke står i forhold til kostnadene.
3.3 Forholdet til næringspolitikken
Legemiddelpolitikken er ikke bare styrt av faglige hensyn, etterspørsel og politiske prioriteringer. Den er også tilbudsstyrt eller tilbyderstyrt. I økende grad fremmer legemiddelprodusenter og andre kommersielle interessenter synet på at en i legemiddelpolitikken også skal ta hensyn til rammevilkårene for industrien når en skal avgjøre spørsmål om for eksempel legemiddelpriser og refusjon.
Dette synet har kanskje særlig vunnet frem i USA og EU, og i andre land med stor farmasøytisk industri. Disse landenes vektlegging av den industripolitiske siden av legemiddelpolitikken har trolig sammenheng med hva landene får tilbake i form av arbeidsplasser, skatteinntekter, viktige kompetansemiljøer etc. Norge kan ikke vise til den samme omfattende legemiddelindustri som land som for eksempel USA, England, Sveits og Sverige. Norsk legemiddelpolitikk har således først og fremst vært utformet for å tilgodese legemiddelbrukerne og holde en forsvarlig kontroll med det offentliges utgifter.
3.4 Departementets vurdering
Sosial- og helsedepartementet mener at målsettingene for legemiddelpolitikken bør videreføres. Departementet vil understreke at tilgjengelighet til legemidler på landsbasis og kvalitet og sikkerhet i legemiddelomsetningskjeden fremdeles må være de overordnede helse- og legemiddelpolitiske mål. Målsetningene bør nås på en mest mulig kostnadseffektiv måte.
Tilgjengelighet til viktige legemidler må i størst mulig grad være uavhengig av pasientenes betalingsevne. Sosial- og helsedepartementet slutter seg til Grundutvalgets forslag til målsetninger for refusjonspolitikken.
4 Sentrale rammevilkår for omsetning av legemidler
4.1 Legemiddelomsetningskjeden - helse og handel
Forsyningen av legemidler frem til publikum involverer både private og offentlige aktører, blant annet legemiddelprodusenter, parallellimportører, legemiddelgrossister, apotek, sykehus og andre helseinstitusjoner. Kjeden omtales gjerne som legemiddelomsetningskjeden.
Myndighetenes reguleringer i legemiddelomsetningskjeden kan ha konsekvenser for alle leddene, men ofte på ulik måte. Måten myndighetene regulerer prisen på legemidler, kan for eksempel ha konsekvenser for tilgjengeligheten til legemidlene, og for prisen forbrukerne må betale. Dette skyldes at valg av prisreguleringsmodell, det vil si om man regulerer for eksempel grossistenes innkjøpspris eller apotekets innkjøpspris, påvirker forhandlingsstyrken og de endelige omsetningsmarginene til de ulike omsetningsleddene.
Omsetning av legemidler omfatter både helsepolitikk og næringspolitikk. Ett av de private aktørenes mål er å sikre forrentningen på investert kapital. Helsemyndighetenes utfordring er å forsøke å finne en politikk som gir befolkningen i alle deler av landet rimelig lett og sikker adgang til effektive og høyverdige legemidler til lavest mulig pris, og sikre høy faglig kvalitet og standard i legemiddelomsetningen, samtidig som hensynet til aktørene i markedet ivaretas i nødvendig grad.
4.2 Prisregulering av legemidler
4.2.1 Industri- og apotekfremstilte legemidler
Den altoverveiende del av legemidlene på markedet er fremstilt av farmasøytisk industri som såkalte farmasøytiske spesialpreparater. Disse selges av apotekene til publikum, normalt uten at apoteket har brutt pakningen. Noen legemidler fremstilles imidlertid for kunden i apoteket, eller i spesielle produksjonsapotek for salg gjennom andre apotek. Slike apotekfremstilte legemidler dekker publikums behov for legemidler som er dårlig egnet for industriell produksjon eller som farmasøytisk industri ikke tilbyr av andre årsaker.
Både reseptpliktige industrifremstilte og apotekfremstilte legemidler er gjenstand for offentlig maksimalprisregulering. Prisreguleringen av de apotekfremstilte legemidlene drøftes i kapittel 12 om tilvirking av legemidler i apotek. I det følgende gjennomgås enkelte generelle prinsipper, samt prisregulering av de industrifremstilte legemidlene.
4.2.2 Hovedformålene med offentlig prisregulering
Alle land med offentlig finansiert helsevesen praktiserer en eller annen form for regulering eller påvirkning av legemiddelprisene for å holde det offentliges kostnader nede. Dette kan skje gjennom direkte prisforhandlinger mellom industri og myndigheter når det avgjøres om nye legemidler skal tas inn i refusjonsordningen, som for eksempel i Sverige og Danmark. Eller det kan skje ved at legemiddelprisene reguleres direkte av myndighetene før legemidlene tillates solgt slik som i Norge. En form for indirekte prisregulering er når refusjonsordningen utformes med sikte på å gi produsentene insentiv til å holde seg innenfor visse maksimalpriser, for eksempel gjennom referanseprissystemer (som i blant annet Norge, Danmark, Sverige, Tyskland og Nederland). I Norge har offentlig prisregulering på legemidler to hovedformål:
Skjerme staten og forbrukerne mot urimelig høye legemiddelpriser som følge av en del kjente imperfeksjoner og særtrekk ved legemiddelmarkedet.
Å bidra til å stimulere aktørene i legemiddelomsetningskjeden til å sikre best mulig tilgjengelighet til legemidler over hele landet.
Forskrift om prisfastsettelse av legemidler av 16. desember 1994 nr. 1116 trådte i kraft 1. januar 1995. Forskriften omfatter alle reseptpliktige legemidler som omsettes i Norge. Prisforskriften innebærer at før et legemiddel kan omsettes eller bringes i handelen, skal dets maksimale utsalgspris fra apotek (AUP) fastsettes. Dette skjer i praksis ved at Statens legemiddelkontroll fastsetter den maksimale innkjøpspris apotekene kan betale grossisten for et legemiddel, det vil si AIP-prisen (apotekets innkjøpspris). Apotekenes avansesatser fastsettes av Sosial- og helsedepartementet etter råd fra Statens helsetilsyn. På grunnlag av fastsatt maksimal AIP og faste satser for apotekavanse, beregner Statens legemiddelkontroll den maksimale apotek utsalgspris (AUP). Dagens regulering av maksimalpriser for legemidler på AIP-nivå (i tillegg til AUP-reguleringen) sikrer apoteket faste og forutsigbare innkjøpspriser fra grossistene for de legemidler som etterspørres.
Figuren nedenfor viser legemiddelomsetningskjeden og dagens prisreguleringsmodell. Det fremgår av figuren at grossistenes innkjøpspris fra produsent (GIP) ikke er underlagt regulering.
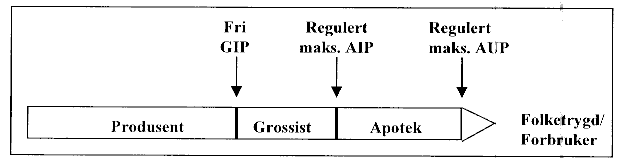
Figur 4.1 Legemiddelomsetningskjeden og dagens prisreguleringsmodell.
4.2.3 Prisregulering og leveringsplikt
4.2.3.1 Målet om god tilgjengelighet til legemidler
I hele etterkrigstiden har det vært en sentral helsepolitisk målsetting at legemidler skal være tilgjengelige for befolkningen uavhengig av bosted. Denne målsettingen gjelder fremdeles, jf. St meld nr 41 for 1987-88 «Helsepolitikken mot år 2000», og vedlegg til St prp nr 1 for 1995-96 «Gjennomgang av legemiddelfeltet».
Både grossister og apotek har såkalt leveringsplikt, det vil si et krav på seg til å skaffe de legemidler som etterspørres innen rimelig tid. Leveringsplikten må ses i nær sammenheng med prisreguleringspolitikken på legemiddelområdet.
4.2.3.2 Grossistenes leveringsplikt
Det er et krav at dagens legemiddelgrossister må kunne levere et hvilket som helst legemiddel med markedsføringstillatelse som etterspørres ved apotek hvor som helst i landet i løpet av 24 timer, jf. forskrift om grossistvirksomhet med legemidler av 21. desember 1993 nr. 1219. I områder med vanskelige kommunikasjonsforhold skal leveranse som hovedregel skje innen 48 timer. Grossisten behøver ikke lagerføre ethvert legemiddel som kan tenkes etterspurt, men har avtaler om levering med produsentene. Leveringsplikten bidrar til å sikre tilgangen til legemidler på landsbasis ved at landets apotek kan kreve levering også av legemidler med liten omsetning og som gir beskjedne økonomiske marginer for grossisten. Uten et slikt krav ville grossisten gitt lavere prioritet til legemidler med lavt omsetningsvolum og lav pris, spesielt i geografisk vanskelig tilgjengelige strøk.
Grossistenes leveringsplikt forhindrer etablering av grossister som bare betjener regionale markeder eller som bare leverer de mest lønnsomme deler av varespekteret. Det er mulig at kostnadene ved grossistvirksomhet kunne bli lavere i visse deler av landet dersom kravet til leveringsdyktighet for alle legemidler på landsbasis ble frafalt. Sosial- og helsedepartementet legger i denne sammenheng størst vekt på at legemidler skal være tilgjengelige på landsbasis, og foreslår derfor i tråd med flertallet i Strømutvalget å videreføre grossistenes leveringsplikt. Kravet til leveringsplikt for grossistene må også ses i sammenheng med tilsvarende krav for apotekene.
4.2.3.3 Apotekenes leveringsplikt
Som en parallell til grossistenes leveringsplikt er apotekene pålagt en plikt til å forhandle alle legemidler som er tillatt solgt her i landet. Dersom en lege rekvirerer et legemiddel som apoteket ikke har på lager eller som sjelden etterspørres, plikter apoteket allikevel å skaffe preparatet til veie. Uten et slikt krav om leveringsplikt kunne tilgjengeligheten til visse legemidler svekkes, spesielt i spredtbygde strøk. Apoteket har dessuten enerett på utlevering av legemidler til det alminnelige publikum utenom institusjon, slik at andre aktører er avskåret fra å levere legemidler dersom apoteket ikke gjør det.
Sosial- og helsedepartementet ser på denne bakgrunn apotekenes leveringsplikt som en sentral forutsetning for å nå den helsepolitiske målsettingen om lik tilgjengelighet til legemidler over hele landet.
4.2.4 Strømutvalgets forslag og høringsinstansenes syn
Flertallet i Strømutvalget foreslo at prisene på reseptbelagte legemidler skulle reguleres på GIP-nivå (grossistenes innkjøpspris), isteden for på AIP-nivå (apotekenes innkjøpspris), i tillegg til på AUP-nivå (apotekets utsalgspris). Det er imidlertid inntatt særmerknader fra flere av medlemmene som ikke deler flertallets syn i dette spørsmålet.
Flertallets forslag får en viss støtte av høringsinstansene til NOU 1997:6, men det er få instanser som går dypt inn i spørsmålet om prisregulering.
4.2.5 Departementets vurdering og forslag til tiltak
Hensikten med prisregulering er først og fremst å skjerme pasienter og folketrygden mot unødig høye legemiddelpriser. Det taler for at maksimalprisen for legemidler bør fastsettes nærmest mulig sluttbruker, det vil si en regulering av apotekenes maksimale utsalgspris. Dette innebærer at omsetningsleddene, det vil si produsenter, grossister og apotek, må fordele legemidlenes maksimale utsalgspris fra apotek seg imellom. De kan således ikke kreve at sluttbruker betaler en høyere pris enn den myndighetene har fastsatt med tanke på å øke egen inntjening. Dersom myndighetene regulerer apotekenes innkjøpspris, uten å sette begrensninger for AUP, kunne apotekeren kreve kunden for den pris apotekeren selv bestemte. Tilsvarende vil både grossister og apotek kunne belaste kunden for høyere omsetningsmarginer, dersom myndighetene kun regulerte GIP. Likeledes ville en full avvikling av offentlig prisregulering komme i konflikt med målsettingen om å skjerme sluttbruker mot høye priser.
På bakgrunn av et ønske om å skjerme sluttbrukerne best mulig mot urimelige høye legemiddelpriser vil Sosial- og helsedepartementet opprettholde dagens ordning med regulering av apotekenes maksimale utsalgspris. I tillegg anser departementet at dagens prisregulering på AIP-nivå må opprettholdes. Ut fra en vurdering av omsetningsleddenes pålagte leveringskrav og partenes forhandlingsstyrke, mener departementet at en slik løsning best ivaretar hensynet til at legemidler fortsatt skal være tilgjengelige over hele landet.
Departementet ser det imidlertid som nødvendig med en løpende vurdering av markedsbetingelsene i legemiddelmarkedet, og vil vurdere både prisreguleringsmodell og kravet om grossistenes og apotekenes leveringsplikt på nytt tre år etter at den nye apotekloven har trådt i kraft, etter at man har vunnet tilstrekkelig erfaring med de nye rammevilkårene for apotekvesenet.
4.3 Aktørene i grossistmarkedet for legemidler
4.3.1 Fullsortiments- og segmentgrossister
Norge har tre fullsortiments legemiddelgrossister. Norsk Medisinaldepot AS (NMD) er landets største grossist og distributør av legemidler og helsepleiemateriell. Hovedkundene er norske apotek og sykehus. NMD ble etablert som et statsselskap i 1957 og fikk samtidig enerett på engrosomsetning av legemidler i det norske markedet. Eneretten ble avviklet 1. januar 1994 som følge av EØS-avtalen. Etter dette har to nye fullsortiments grossister etablert seg, Holtung AS og Tamro Distribution AS. Norsk Medisinaldepot hadde i 1997 en markedsandel på 72,4 prosent, mens Holtung AS og Tamro hadde markedsandeler på henholdsvis 20,6 prosent og 7,0 prosent. Norsk Medisinaldepot driver sin engrosvirksomhet kun i Norge, mens Holtung og Tamro også opererer i andre nordiske land.
På legemiddelmarkedet er det også enkelte grossister som betjener spesielle markedssegmenter og som har et begrenset sortiment. En slik grossist er Veterinærmedisinsk Oppdragssenter AS (VESO) som driver omsetning av veterinære vaksiner, og EuroPharma AS som driver med det samme. I tillegg kan firmaer med importtillatelse for legemidler være grossist for sitt eget sortiment. Tilsvarende kan legemiddeltilvirkere drive grossistvirksomhet for de legemidler som tilvirkergodkjenningen gjelder for.
Grossister kan som hovedregel selge legemidler bare til apotek. Gjennom legemiddeloven § 15 er imidlertid departementet gitt fullmakt til å godkjenne salg fra grossist direkte til sykehus og visse andre sluttbruker, uten å gå veien om apotek. Slike direkteleveranser er begrenset til bestemte varegrupper som ifølge lovens system skal fastsettes etter departementets nærmere bestemmelser.
4.3.2 Salg av statsaksjer i Norsk Medisinaldepot AS
I NOU 1997:6 uttalte Strømutvalget at «...utvalget ser ingen helseøkonomiske grunner til at staten skal eie den ene av dagens tre grossister».
I St prp nr 63 for 1996-97 ba Regjeringen om fullmakt fra Stortinget til at Sosial- og helsedepartementet kunne gå i gang med forhandlinger med sikte på helt eller delvis salg av statens eierinteresser i Norsk Medisinaldepot. Stortinget sluttet seg til Regjeringens forslag, og ba Regjeringen komme tilbake til Stortinget for endelig godkjenning av avtalen, jf. vedtak av 20. juni 1997 (Innst S nr 296 for 1996-97).
I St prp nr 65 for 1997-98 ba regjeringen om fullmakt fra Stortinget til endelig salg av inntil 66 prosent av statens andeler i Norsk Medisinaldepot. Salget skulle skje innenfor de vilkår Stortinget tidligere hadde lagt til grunn:
Staten beholder en eierandel på minimum 34 prosent.
Bredest mulig spredning av eierskapet på norske hender.
Oppnå best mulig pris.
Sikre kjøpere/alliansepartnere som bidrar til økt konkurranse og effektiv ressursbruk i legemiddelmarkedet.
Styrke bedriftens markedsposisjon og trygge arbeidsplassene.
Sikre forsvarlig beredskap av legemidler.
Stortinget sluttet seg til Regjeringens forslag i Innst S nr 252 for 1997-98. Sosial- og helsedepartementet arbeider for tiden med de nødvendige forberedelser til et salg på grunnlag av de fullmakter Stortinget har gitt. Departementet tar sikte på å gjennomføre aksjesalget i Norsk Medisinaldepot i løpet av våren 1999.
4.3.3 Departementets vurdering og forslag til tiltak
Etter departementets vurdering fungerer grossistleddet i legemiddelomsetningskjeden bra etter reformen den 1. januar 1994 med fri etablering i grossistleddet. To nye fullsortimentsgrossister på markedet har ledet til konkurranse, som trolig har bidratt til bedre service og lavere priser i grossistleddet. Som Strømutvalget (NOU 1997:6) ser departementet ingen tungtveiende grunner til å endre dagens ordning med fri etablering av grossister.
Departementet ser derimot et behov for en noe større adgang for grossister enn i dag til å levere legemidler direkte til sluttbruker utenom apotek. For en del profesjonelle sluttbrukere kan kjøp fra apotek etter omstendighetene være unødig tungvint. Dette gjelder brukere som legekontorer, kommunale og fylkeskommunale helseinstitusjoner samt andre profesjonelle brukere som ikke har behov for samme type farmasøytiske kompetanse som publikum har behov for, og som apotekene er særlig innrettet på å dekke. Profesjonelle brukere av legemidler har spesielle behov når det gjelder farmasøytisk bistand og service, bestillingsrutiner, innkjøpsbetingelser m.v. Spesialiserte enheter på grossistnivå med hele eller deler av landet som marked kan være bedre egnet til å dekke profesjonelle brukeres behov enn lokale apotek, som primært skal dekke det alminnelige publikums behov. Etter departementets syn er det et visst rom for utvikling av enheter på grossistnivå som innretter seg mot profesjonelle legemiddelbrukere. Det kan gi denne kategorien brukere et bedre og mer kostnadseffektivt tilbud enn i dag.
For å legge til rette for en slik utvikling foreslås det enkelte endringer i legemiddelloven § 15 tredje ledd (jf. kapittel 14.10 om endringer i andre lover). Endringen innebærer at departementet får fullmakt til å bestemme at grossister kan selge nærmere angitte legemidler til profesjonelle sluttbrukere som lege- og tannlegekontorer samt offentlige og offentlig godkjente helseinstitusjoner, i tillegg til de sykehus og laboratorier o.l. som grossister i dag har anledning til å selge legemidler direkte til utover salget til apotek. Med hensyn til hvilke legemidler som skal omfattes av ordningen, tilsier faglige grunner at det sondres mellom de forskjellige kategorier legemiddelbrukere etter deres legemiddelfaglige innsikt. Departementet tar derfor sikte på å utvide vesentlig det relativt beskjedne legemiddelsortimentet som sykehus med egen farmasøytisk ekspertise i dag har adgang til å kjøpe direkte fra grossist.
Selv om strukturelle forhold taler for at profesjonelle legemiddelbrukere i større grad får anledning til å kjøpe legemidler direkte fra aktører som har innrettet seg mot å dekke deres særlige behov, kan distriktshensyn trekke i motsatt retning. Dette vil være tilfelle hvis økt konkurranse i det profesjonelle markedet fører til at distriktsapotek mister leveranser til institusjonelle kunder som apoteket er avhengig av for å overleve økonomisk. Departementet vil nøye vurdere distriktsvirkningen før en går til en eventuell utvidelse av grossistenes adgang til å selge legemidler direkte til profesjonelle sluttbrukere innenfor kommunehelsetjenesten. Hvis adgangen blir utvidet, er det dessuten naturlig at apotek blir satt i stand til å konkurrere om de samme profesjonelle legemiddelbrukerne gjennom utvidet adgang til legemiddelsalg til slike kunder via postforsendelser, se nærmere kapittel 11.1.1.4.
4.4 Parallellimport av legemidler
4.4.1 Bakgrunn og begreper
Parallellimport er innførsel av varer, i denne sammenheng legemidler, utenom produsentens eller importørens ordinære forhandlernett. Slik import tar sikte på videresalg i konkurranse med de etablerte forhandlerne. Betegnelsen parallell er uttrykk for at samme legemiddel importeres via to parallelle kanaler. Det direkteimporterte legemidlet er produsert på moderfabrikken og selges direkte derfra til Norge gjennom en fast representant. Det parallellimporterte preparatet kan være produsert i moderfabrikken og solgt til et annet land, hvor det så kjøpes av en representant og importeres til Norge. Vanligere er det at legemidlet produseres i et annet land, enten i datterfabrikk eller på lisens, og kjøpes derfra av en representant og importeres til Norge. Det økonomiske grunnlaget for parallellimport er at prisen på samme produkt kan variere mellom europeiske land. Parallellimportører oppnår salgsgevinster ved å kjøpe legemidler i land med lave priser og selge dem i markeder hvor prisen på det samme legemidlet er høyere. Det er først og fremst de mestselgende, patenterte legemidlene som parallellimporteres. Dette fører til at preparater med stort salg ofte markedsføres av flere importører, og det blir en viss priskonkurranse.
Legemiddelindustrien i Norge har gått til dels sterkt ut mot parallellimport. Blant annet har det vært fremholdt at parallellimport av legemidler øker faren for omsetning av forfalskede og potensielt skadelige produkter. Ettersom det er de direkteimporterende, nasjonale legemiddelfirmaene som bærer kostnadene for produktinformasjon og reklame, fremholdes det at parallellimportørene «skummer fløten» av direkteimportørenes arbeid. Det er allikevel legemiddelfirmaets aksjonærer som nyter godt av størstedelen av inntektene, ettersom alle de impliserte legemidlene stammer fra samme firma.
Parallellimport er en følge av EØS-regelverket. Prinsippet om fri flyt av varer og tjenester betyr at medlemslandene ikke lenger kan forby eller begrense adgangen til å drive parallellimport av andre grunner enn vernet av menneskers og dyrs helse. Parallellimport av ikke patentbeskyttede legemidler ble tillatt i Norge fra og med 1994. Fra og med 1995 er parallellimport tillatt også for de patentbeskyttede legemidlene. De første parallellimporterte preparatene kom på det norske markedet i mai 1995. Sosial- og helsedepartementet gir her en nærmere omtale av parallellimportørenes virksomhet og hvilke sentrale krav m.m. myndighetene stiller til kvalitetssikring av slike legemidler.
4.4.2 Parallellimportørene og deres omsetning
Det er pr. november 1998 ti firmaer som er registrert som rene parallellimportører av legemidler i Norge. Importørene selger videre til de norske legemiddelgrossistene, som står for videresalg til apotekene. I 1996 utgjorde parallellimporterte preparater om lag to prosent av totalomsetningen for legemidler. I 1997 hadde andelen økt til ca. seks prosent.
Selv om parallellimportørenes markedsandeler totalt sett er forholdsvis lave, vil markedsandelen være vesentlig høyere på utvalgte og viktige legemiddelområder. I tillegg kan parallellimport bidra til økt priskonkurranse i forhold til direkte importerte legemidler. For å unngå tap av markedsandeler må leverandørene av disse fortløpende vurdere sin prispolitikk. Effektiv priskonkurranse på patenterte legemidler vil gi vesentlige innsparinger i trygdens utgifter til legemidler. Det vises til omtale av referanseprissystemet i St prp nr 1 for 1998-99 (Folketrygden).
Det må søkes om markedsføringstillatelse for hvert parallellimporterte preparat som ønskes solgt i Norge. Ett preparat, importert fra to forskjellige land, krever totalt to markedsføringstillatelser.
Det er gitt over 500 markedsføringstillatelser for parallellimporterte legemidler hittil, fordelt på over 200 i henholdsvis 1995 og 1996, og om lag 100 i 1997. I 1998 er det pr 30. oktober gitt markedsføringstillatelse for 44 parallellimporterte preparater. Av de drøyt 500 preparatene er det bare i overkant av 300 som er kommet på markedet. Dette skyldes at det kreves markedsføringstillatelse for hvert enkelt leverandørland dersom et preparat produseres i flere land. Produsentene søker for sikkerhets skyld om tillatelse til å importere et preparat fra flere land, for å sikre seg mot leveringsvansker og nasjonale prissvingninger. I praksis benyttes ikke alle tillatelsene samtidig.
4.4.3 Kvalitetssikring av parallellimporterte legemidler
4.4.3.1 Kvalitetssikring før tildeling av markedsføringstillatelse
For at et parallellimportert legemiddel skal få markedsføringstillatelse i Norge, må det foreligge dokumentasjon på at preparatet har markedsføringstillatelse i eksportlandet, og at det tilsvarer et direkteimportert preparat fra samme produsent med gyldig markedsføringstillatelse i Norge. Hvis ikke slik dokumentasjon kan fremskaffes, vil søknaden om markedsføringstillatelse bli avslått. Gjennomgang av denne dokumentasjonen utgjør den viktigste kvalitetskontrollen av et parallellimportert legemiddel. Av spesielle forhold som skal dokumenteres omfattes:
Bekreftelse på at preparatet har markedsføringstillatelse i eksportlandet og angivelse av hvem som innehar denne.
Hvem som er tilvirker og eventuelt ansvarlig for import til eksportlandet.
Holdbarhetstid og holdbarhetsbetingelser for både parallell- og direkteimportert preparat.
Eventuelle forskjeller i utseende fra direkteimportert preparat.
Beskrivelse av eventuell ometikettering og ompakking, herunder hvem som foretar dette og hvor det foregår.
Forslag til pakningsmateriale som skal benyttes i Norge, og prøve på pakningsmateriale som benyttes i eksportlandet.
I tillegg innhenter Statens legemiddelkontroll oversikt over preparatets sammensetning fra legemiddelmyndighetene i eksportlandet. Ved mindre avvik i sammensetning mellom parallellimportert og direkteimportert preparat, vil det bli vurdert om preparatene er terapeutisk likeverdige. Hvis det parallellimporterte preparatet vurderes som terapeutisk likeverdig med det direkteimporterte, vil markedsføringstillatelse bli gitt, selv om sammensetningen er noe forskjellig. Dette kan gjelde egenskaper som smak, farge, konsistens og annet. Reglene for parallellimport sikrer likevel at begge preparatene tilfredsstiller kravene til kvalitet, sikkerhet og effekt. Trenden er harmonisering av produksjon og godkjenning for det europeiske markedet, slik at ytre ulikheter av denne typen etterhvert vil bli mindre.
For biologiske preparater stilles det spesielt strenge krav til dokumentasjon av likeverdighet. Med biologiske legemidler menes legemidler med virkestoff fremstilt fra humane eller animalske kilder, eller fremstilt ved dyrking av mikroorganismer eller cellekulturer. Kvaliteten på slike legemidler kan i større grad enn andre legemidler påvirkes av mindre forskjeller i fremstillingsmetode og kontrollprosedyre. Det kreves derfor spesifisert dokumentasjon vedrørende produksjonssted, fremstillingsmetode og kontrollprosedyrer.
4.4.3.2 Krav til merking
Kontroll av innsendt forslag til merking er en viktig del av kvalitetskontrollen. Dersom innsendt forslag ikke tilfredsstiller de krav som er satt, vil det aktuelle preparat bli nektet solgt i Norge. Parallellimport vil bli godkjent selv om pakningsstørrelser og pakningstyper avviker fra det direkteimporterte, så lenge det ikke kommer i konflikt med dosering og indikasjon godkjent i Norge.
Det er de samme merkingsreglene som gjelder for parallellimporterte som for direkteimporterte preparater. Legemidler skal merkes i henhold til EUs merkingsdirektiv. Formålet med direktivet er å sikre høy sikkerhet for forbrukerne. Legemidlene skal kunne brukes på riktig måte på grunnlag av fullstendige og entydige opplysninger. Opplysningene skal være lett leselige, klart forståelige og ikke kunne slettes. Opplysningene skal gis på landets offisielle språk. De kan i tillegg gis på andre språk, forutsatt at informasjonen er identisk. For parallellimporterte preparater kan dette gjøres ved at utenlandsk pakning påføres en etikett med de påkrevde opplysninger på norsk. Det kan kreves tilleggsmerking av parallellimporterte legemidler for å opplyse særskilt om eventuelle forskjeller i forhold til direkteimporterte.
I et brev fra Norges Apotekerforening til Statens helsetilsyn av 10. juli 1998 klages det på at inner- og ytterpakningen for tre parallellimporterte legemidler har vært merket med forskjellige navn. Statens legemiddelkontroll har på bakgrunn av henvendelsen foretatt nødvendige forbedringer, og strammet inn og klargjort hvilke retningslinjer som gjelder for slik merking.
Det parallellimporterte preparatet vil i utgangspunktet få samme holdbarhetstid som den som er gjeldende i eksportlandet.
4.4.3.3 Kontroll etter tildeling av markedsføringstillatelse
Da de første parallellimporterte legemidlene ble markedsført i Norge i 1995, ble disse underkastet laboratoriekontroll på lik linje med de direkteimporterte. Etter en prøveperiode på om lag et halvt år, viste kontrollen følgende:
Parallellimporterte preparater har samme kjemiske og farmasøytiske kvalitet som direkteimporterte.
De største feilkildene ved parallellimporterte preparater er pakning, ompakning, etikettering og tekster.
Etter prøveperioden ble det bestemt å innføre en forenklet førstegangskontroll av de parallellimporterte preparatene, for deretter å samkjøre etterkontrollen med det direkteimporterte preparatet. Dette systemet benyttes i dag.
Like etter markedsføring blir det derfor utført en forenklet laboratoriekontroll, en såkalt sikkerhetskontroll, av det parallellimporterte preparatet. Denne kontrollen innebærer analyse av identitet og mengde av virkestoff samt en undersøkelse av pakning og merking. Hvis produsentens metode for bestemmelse av mengde av virkestoff er ressurskrevende, vil i mange tilfeller en enklere analysemetode velges i denne fasen. Ved siden av førstegangskontrollen blir det utført en fullstendig laboratorieanalyse hvert femte år. Etterkontrollen blir samkjørt med det direkteimporterte preparatet, slik at første fullstendige analyse av det parallellimporterte preparatet vil skje fem år etter siste kontroll av det direkteimporterte. Fordelene ved dagens forenklede kontrollopplegg er:
Bedre mulighet for sammenligning av kvalitet på direkteimportert og parallellimportert preparat.
Arbeidsbesparelse ved gjennomføring av kompliserte analysemetoder og rapportskriving.
Når det gjelder innholdet i pakningen, har feilfrekvensen vært omtrent den samme for parallellimporterte som for direkteimporterte legemidler. I tillegg er det funnet sporadiske feil ved ompakking eller ometikettering av parallellimporterte preparater.
Parallellimportørene er pålagt å holde seg informert om eventuelle endringer av sine preparater, og skal holde Statens legemiddelkontroll løpende orientert. Dette følges i varierende grad opp av importørene. Statens legemiddelkontroll får alltid melding i de tilfellene der det parallellimporterte preparatet får ny markedsføringstillatelse i eksportlandet, og i disse tilfellene tas det rede på hvilken endring som er foretatt. Endringer som ikke fører til ny markedsføringstillatelse, har Statens legemiddelkontroll derimot ikke alltid oversikt over. De forskjellige EØS-landene har ulik praksis når det gjelder hvilke endringer som krever ny tillatelse. Legemiddelmyndigheten i eksportlandet har imidlertid oversikt over alle endringer, og Statens legemiddelkontroll kan få opplysningene derfra ved behov.
Når Statens legemiddelkontroll godkjenner endringer av preparatomtalen for det direkteimporterte preparatet, vil parallellimportøren umiddelbart få tilsendt den nye preparatomtalen med beskjed om å ta i bruk denne.
4.4.4 Departementets vurdering og forslag til tiltak
Den generelle utviklingen som har funnet sted som følge av EØS-tilpasningen med økende antall beslektede, synonyme eller parallellimporterte legemidler, har gjort legemiddeltilbudet mer sammensatt og krevende å holde oversikt over for både leger og pasienter. Statlige legemiddelmyndigheter har imidlertid ikke holdepunkter for at den økte andelen parallellimporterte legemidler, eller for den saks skyld synonympreparater, fører til økt feilbruk av legemidler. Det har blitt hevdet at samme legemiddel med ulikt utseende vil kunne redusere pasientens etterlevelse av legens forordning, for eksempel ved at en pasient som har beholdning av både gamle og nye medisiner, tar dobbel dose. Slike forhold er apotekene oppmerksomme på, og de driver en omfattende veiledning av pasientene ved utlevering av medisiner.
Det er imidlertid nødvendig at hele behandlingskjeden bidrar til å sikre riktig bruk av legemidler. Gjennom forskrivning av legemidler har legen en helt sentral informasjonsrolle. Ved innføring av fastlegeordningen vil det kunne etableres tettere og mer faste relasjoner mellom lege og pasient. Hjemmesykepleien kan for eksempel også bidra til å etterse at pasienter tar sine medisiner som forskrevet. Apotekene kan bidra gjennom fortsatt kvalitetssikring av legenes forskrivning og god informasjon til pasientene.
Sosial- og helsedepartementet ser parallellimport som et viktig virkemiddel for å skape økt priskonkurranse i legemiddelmarkedet. Dette kan komme forbrukere, offentlige helseinstitiusjoner og folketrygden til gode gjennom lavere utgifter til legemidler. Departementet ser imidlertid at parallellimport kan gi et mer uoversiktlig legemiddelmarked. For å motvirke risikoen for feilbruk som følge av parallellimport vil Statens legemiddelkontroll fortsatt stille strenge krav til kvalitetssikring av parallellimporterte legemidler, og om nødvendig skjerpe disse. I tillegg pålegges apotekene i forslag til ny apoteklov å bidra til at pasienten får tilstrekkelig informasjon om legemidlet til at det kan brukes riktig, se nærmere kapittel 11.2.1. Det vises også til kapittel 5.1.3 med omtale av referanseprissystemet.
4.5 Reseptfrie legemidler og egenomsorg
4.5.1 Særlige trekk ved reseptfrie legemidler
Reseptfrie legemidler er legemidler som riktig brukt er trygge ved behandling av symptomer og tilstander som egner seg for egenomsorg. Eventuell risiko knyttet til bivirkninger og feilbruk, samt muligheten for å dekke over symptomer på alvorlig sykdom, trekkes inn i vurderingen av reseptfrihet.
Faglige råd om håndtering av lettere plager vil hjelpe kunden til å velge riktig behandling. Dersom behandling med legemidler velges, kan råd om bruken være avgjørende for et godt resultat. God egenomsorg betyr en reell avlastning for øvrig helsetjeneste. Apotekene er en viktig møteplass i forbindelse med egenomsorg.
4.5.2 Salg av reseptfrie legemidler utenom apotek
Reseptfrie legemidler selges både gjennom landets 350 apotek og filialapotek, samt gjennom de ca. 1.290 medisinutsalgene som er underlagt apotekene. I 1996 utgjorde salg av reseptfrie legemidler omlag 9,1 prosent av apotekenes omsetning, eller ca. 740 millioner kroner. Strømutvalget drøftet i NOU 1997:6 hvorvidt reseptfrie legemidler skulle kunne selges gjennom andre kanaler enn gjennom apotek. Utvalget delte seg på midten i spørsmålet om hvordan dette kunne gjennomføres. Den ene halvparten mente at reseptfrie legemidler, som myndighetene finner kan selges fritt uansett pakningsstørrelse, skulle tillates solgt utenom apotek. Den andre halvparten mente at bare legemidler som rette fagmyndighet finner kan selges reseptfrie uansett pakningsstørrelse og indikasjon, burde tillates solgt utenom apotek, forutsatt at de ikke kan misbrukes, ikke forårsake alvorlige interaksjoner og/eller bivirkninger og ikke krever kvalifisert veiledning.
Strømutvalget kom i liten grad inn på hvilke former for oppmykning som faktisk var aktuelle. Ved salg av reseptfrie legemidler gjennom kanaler som bensinstasjoner og dagligvareforretninger vil en oppnå langt større tilgjengelighet enn i dag. Alternativt kan det tenkes mindre vidtrekkende alternativer, som for eksempel salg gjennom forretninger som oppfyller vilkår til sikker legemiddelhåndtering, veiledning av kunder m.m. etter krav som i dag stilles til medisinutsalg.
4.5.3 Høringsinstansenes syn
Høringsinstansene til NOU 1997:6 uttrykte ulike syn i spørsmålet om reseptfrie legemidler skal kunne selges utenom apotek. Et klart flertall av faginstansene var skeptiske til en slik liberalisering. Statens legemiddelkontrollhadde ikke motforestillinger mot salg av reseptfrie legemidler utenom apotek. Forutsetningen var at slike legemidler må være reseptfrie uansett pakningens størrelse.
Giftinformasjonsentralenla et meget omfattende statistikk- og analysemateriale til grunn for sin høringsuttalelser. Blant annet ble det presentert statistikk over henvendelser til Giftinformasjonen etter årsak, aldersgrupper, forgiftningsgrad m.v. Giftinformasjonssentralen var betenkt over at legemidler eventuelt skal selges i dagligvareforretninger og på bensinstasjoner etc. og bli liggende ute i lett tilgjengelige hyller hvor det er et minimum av betjening til å føre kontroll. Det ble fremholdt at en økning i feilbruk, misbruk og antall forgiftninger av legemidler vil føre til økt belastning og økte utgifter i helsevesenet.
I sin høringsuttalelse til Sosial- og helsedepartementets høringsnotat om ny apoteklov skriver Norges Colonialgrossisters Forbundat reseptfrie legemidler generelt bør anses som vanlig handelsvare:
«Påstanden om at dagligvarebutikker ikke har kompetanse til å veilede for eksempel astmapasienter om at bruk av acetylsalisylsyre (Dispril) kan utløse astmautslag, er ikke i utgangspunktet korrekt fordi slike medikamenter i dagligvarebutikk kan tenkes solgt på en slik måte at problemet ivaretas (særskilt gruppering, indikasjoner etc.). (...) Vi har full forståelse for at legemidler ikke kan anses som handelsvarer på lik linje med andre nødvendighetsgoder som for eksempel matvarer pga. helsekonsekvenser og økonomiske konsekvenser for forbrukerne og for å sikre kontroll, men mener reseptfrie legemidler generelt bør anses som vanlig handelsvare».
4.5.4 Departementets vurdering og forslag til tiltak
For å øke folks muligheter til egenomsorg er en rekke reseptpliktige legemidler de senere år blitt unntatt fra reseptplikt i små pakninger, eventuelt for bestemte indikasjoner. Det gjelder for eksempel en del smertestillende midler, nesedråper og allergimidler. For disse legemidlene er risikoen knyttet til bivirkninger og feilbruk noe større enn for legemidler som er helt reseptfrie, men fortsatt liten.
Sosial- og helsedepartementet anser tilgjengeligheten til reseptfrie legemidler gjennom apotek og medisinutsalg som forsvarlig. Mer tilfeldig tilgjengelighet til reseptfrie legemidler kan bidra til å redusere forbrukernes respekt og forsiktighet ved bruk av reseptfrie legemidler. Departementet vil i den sammenheng understreke at også reseptfrie legemidler kan ha alvorlige, og endog i spesielle situasjoner livsfarlige bivirkninger. Videre har mange kunder behov for sakkyndig veiledning i apoteket for å sikre riktig bruk. Dette behovet forsterkes ved at pasienten ikke har fått slik informasjon i forbindelse med utstedelse av resept hos lege. Departementet mener at dagligvarebutikker ikke vil kunne ivareta veiledningsoppgaven i forbindelse med reseptfrie legemidler på en like faglig god måte som apotekene.
Departementet er også bekymret over at salg av legemidler utenom apotek kan skape mindre effektive rutiner når det gjelder tilbakekall av medikamenter med feil og mangler. Et stort forhandlernett for reseptfrie legemidler må forventes å gi dårligere oversikt over utsalgsstedene enn i dag.
Sosial- og helsedepartementet vil blant annet på denne bakgrunn ikke foreslå at det gjøres endringer i apotekenes enerett til salg av reseptfrie legemidler. Gjennom den foreslåtte oppmykningen av dagens etableringspolitikk for apotek, vil tilgangen til reseptfrie legemidler bedres. Departementet vil i samarbeid med sine faginstanser vurdere salg av reseptfrie legemidler utenom apotek på nytt tre år etter at en ny apoteklov har trådt i kraft, etter at man har vunnet tilstrekkelig erfaring med det nye apoteksystemet.
4.6 Legemiddellovens regulering av markedsføring
4.6.1 Innledning
Betalingsvilligheten for produkter som hevdes å ha helbredende eller lindrenede effekt må antas å være høy for mennesker som plages av sykdom eller lider av smerter. På den annen side må hensynet til vern om liv og helse ivaretas på en betryggende måte. Myndighetene har således et ansvar for å stille krav til og føre kontroll med kvalitet, effekt og sikkerhet vedrørende produkter som er bestemt for eller utgis for å være medisinsk virksomme.
Hvordan en vare frembys på markedet, kan være avgjørende for om varen skal ansees som legemiddel. Det er bare registrerte legemidler, som oppfyller kravene til kvalitet, sikkerhet og effekt, som kan omsettes lovlig i Norge. Registreringen av legemidlet gir også anledning til å markedsføre preparatet med medisinske påstander. Av hensyn til konsistens i regelverket og ut fra helsepolitiske betraktninger, herunder hensynet til folks helse, vil det være svært uheldig dersom alminnelige handelsvarer skulle kunne markedsføres med påstander om medisinsk effekt.
Ved å gi næringsmidler legemiddellignende form kan man gjennom reklame med medisinske påstander skape urealistiske forventninger hos pasienter og forbrukere om varens helbredende eller forebyggende effekt. Pasienten eller forbrukeren kan bli skadelidende, fordi vedkommende unnlater å oppsøke lege eller slutter å ta sine medisiner i tillit til påstandene.
4.6.2 Historikk
Medisinsk reklame for alminnelige handelsvarer har vært regulert med forbud siden Apotekhandelsplakaten. I avdeling D i plakaten av 26. februar 1954, som omfattet varer som ikke var apotekvarer eller gifter (altså alminnelige varer eller ikke-legemidler), var det blant annet bestemt:
«Disse varer og andre varer som ikke hører inn under foregående avdelinger, må ikke fallbys på en slik måte at de fremtrer som legemidler. De må således ikke ved annonser, sirkulærer eller på annen måte frembys til salg eller anbefales som midler til å forebygge, lindre eller lege sykdom».
Apoteklovkomiteen (se kapittel 9.1.5) uttalte i sin innstilling II fra 1959 vedrørende lov om legemidler at:
«Det må ansees for å være meget uheldig at varer som ikke omsettes som legemiddel reklameres på en slik måte at det fremgår at de har forebyggende eller helbredende virkning mot sykdommer».
Komiteen gikk således inn for å opprettholde forbudet mot medisinsk reklame for ikke-legemidler. Samtidig mente komiteen at det var behov for å styrke håndhevingen av forbudsbestemmelsen med et mer effektivt sanksjonssystem. Komiteen åpnet i loven adgang for å kunne forby ved dom den ulovlig reklamerte varen solgt under samme varebetegnelse. Behovet ble begrunnet med at det i enkelte tilfelle bevisst hadde vært benyttet medisinsk reklame for alminnelige handelsvarer. Ofte oppnådde markedsføreren å gjøre varen kjent som virksom mot sykdom før myndighetene hadde kunnet ta forholdet opp og søke å stanse reklamen. Dessuten forekom det ikke sjeldent at firmaer som hadde fått advarsler og blitt ilagt bøter for ulovlig medisinsk reklame for alminnelige varer, likevel fortsatte, i håp om at det økte salget de kunne oppnå ved medisinske påstander skulle oppveie boten.
4.6.3 Gjeldende rett
4.6.3.1 Innledning
Lov av 4. desember 1992 nr. 132 om legemidler m.v. (legemiddelloven) regulerer tilvirkning og omsetning av legemidler. Legemiddelloven § 2 definerer legemidler:
«Med legemidler forstås i denne lov stoffer, droger og preparater som er bestemt til eller utgis for å brukes til å forebygge, lege eller lindre sykdom, sykdomssymptomer eller smerter, påvirke fysiologiske funksjoner hos mennesker eller dyr, eller til ved innvortes eller utvortes bruk å påvise sykdom».
Annet ledd i § 2 gir Kongen hjemmel til å utferdige forskrifter om hva som skal regnes som legemidler. Som det fremgår av legemiddelloven § 2 er definisjonen av hva som ansees som legemidler meget vidtfavnende. Dette har sammenheng med at det i en slik legaldefinisjon ikke er praktisk mulig å ta utgangspunkt i stoffenes art eller kjemiske og fysiske egenskaper. Et legemiddel defineres således ikke bare ut fra dets beskaffenhet, men hva midlet skal nyttes til vil være et avgjørende moment om det skal regnes som legemiddel i henhold til legemiddelloven § 2. Et viktig moment i denne forbindelse er hvorledes varen frembys. Bringes den i handelen som legemiddel ved å utgis som medisinsk virksom på fysiologiske prosesser hos mennesker eller dyr, så er den underkastet lovens restriksjoner som blant annet omfatter plikten til søke om registrering (markedsføringstillatelse).
Reklame for legemidler reguleres av legemiddelloven som uttrykker i § 19 at «reklame for legemidler skal være nøktern og sann». Videre er det med hjemmel i legemiddelloven utferdiget forskrift av 25. august 1994 nr. 827 om reklame for legemidler med utfyllende regler om innholdet for legemiddelreklamer og kontroll med reklame for legemidler. Overtredelse av reklamebestemmelsene kan medføre reaksjoner fra forvaltningen og/eller forfølges strafferettslig, jf. legemiddelloven § 31 og reklameforskriften § 10.
4.6.3.2 Reklame med medisinske påstander
Som en logisk konsekvens av legemiddellovens definisjon av legemidler i § 2 forbeholder loven adgangen til å anvende medisinske påstander i markedsføring bare for legemidler registrert i henhold til loven. Dette kommer til uttrykk i legemiddelloven § 20, som bestemmer:
«Det er forbudt i reklame eller lignende, ved tekst eller bilder, direkte eller indirekte, å gi uttrykk for at en vare som ikke er legemiddel anbefales som middel til å forebygge, lege eller lindre sykdom, sykdomssymptomer eller smerter eller påvirke fysiologiske funksjoner hos mennesker eller dyr. Når særlige grunner foreligger, kan departementet gjøre unntak fra denne bestemmelse».
Dette er en forbudsbestemmelse gjeldende for varer som ikke er legemidler i en lov som regulerer legemidler. Legemiddelloven § 20 omhandler reklame for varer som selges fra alminnelige salgssteder, men som angis å være legemidler eller tilskrives medisinske virkninger eller egenskaper.
4.6.3.3 Legemiddelloven § 20 - hensyn og formål
Av motivene til legemiddelloven fremgår det at meningen med loven ikke er å skaffe hjemler for kontroll med produksjon og omsetning ut fra andre hensyn enn vern om liv og helse.
Legemiddelloven § 20 er således begrunnet ut fra hensynet til folks helse og den beskyttelsesverdige interessen forbrukerne har i at varer ledsaget med medisinske påstander virkelig svarer til forventningene. Videre er konsistens og logisk sammenheng i lovverket et hensyn av ikke ubetydelig vekt som berettiger forbudet mot medisinsk reklame i legemiddelloven § 20 hensett til definisjonen i legemiddelloven § 2, og det faktum at selv reklame for godkjente legemidler er underkastet regulering og kontroll, jf. legemiddelloven § 19 m.v.
Formålet med legemiddelloven § 20 er å hindre at varer som ikke er legemidler blir bevisst reklamert for med medisinske påstander, slik at disse overfor forbrukerne fremstår som produkter med medisinsk effekt uten at dette er godkjent av fagmyndighetene. For legemiddelloven § 20 er spørsmålet om benyttede medisinske påstander faktiske er sanne, uten relevans.
4.6.3.4 Det materielle innhold i legemiddelloven § 20
Forbudet mot medisinsk reklame i legemiddelloven § 20 omhandler reklame for varer som ikke er legemidler. Varer som faller inn under definisjonen av legemidler i legemiddelloven § 2, omfattes således ikke av legemiddelloven § 20. For et produkt som er legemiddel etter legemiddelloven § 2, må det søkes om markedsføringstillatelse for å kunne bli omsatt lovlig, jf. legemiddelloven § 8.
Videre anvender legemiddelloven § 20 et vidt begrep om reklame. Forbudet i bestemmelsen rammer «reklame eller lignende, ved tekst eller bilder, direkte eller indirekte ...». Alle former for kommersielle markedsføringstiltak som er egnet til å fremme salg, samt andre tiltak som er egnet til å påvirke etterspørselen eller tilbudet av varer, omfattes av bestemmelsen. Det er formålet som har betydning for grensedragningen mellom ytring og reklame. Det vil således inngå i reklamebegrepet i § 20 dersom man benytter seg av henvisning til mer eller mindre vitenskapelige artikler om varens medisinske egenskaper, eller til seminarer, presentasjoner eller lignende hvor det gis slik informasjon. Legemiddelloven § 20 sondrer ikke med hensyn til om reklamen er rettet mot forbrukerne eller fagfolk.
Den reklame som i henhold til legemiddelloven § 20 er ulovlig, må inneholde medisinske påstander. Legemiddelloven § 20 uttrykker kriterier for hva som skal ansees som medisinsk påstand:
«... middel til å forebygge, lege eller lindre sykdom, sykdomssymptomer eller smerter eller påvirke fysiologiske funksjoner hos mennesker eller dyr».
Hovedkriteriet kan sammenfattes i begrepet «påvirke fysiologiske funksjoner», dog er ikke enhver påvirkning relevant. Utvilsomt er det at loven ikke skiller mellom sanne og usanne påstander. Hva som nærmere bestemt betraktes som medisinsk påstand, beror på et faglig skjønn der hensynet til folks helse og helsepolitiske overveielser, herunder påvirkning av ernæringsmessig forhold og legemiddelforbruk i ønsket retning, vil være vektige momenter. Statens legemiddelkontroll har utarbeidet retningslinjer for hva som ansees som medisinske påstander.
Forbudet etter legemiddelloven § 20 er absolutt. Det rammer ulovlig reklame uansett hvilket ledd i omsetningskjeden som står for utgivelse av reklamen.
4.6.3.5 Tilstøtende lovgivning - tilgrensende områder
Næringsmidler reguleres av lov av 19. mai 1933 nr. 3 om tilsyn med næringsmidler m.v. Lovens § 1 gir en definisjon av næringsmidler:
«Med næringsmidler menes enhver mat- eller drikkevare, også drikkevann, og enhver annen vare som er bestemt til å konsumeres av mennesker, unntatt legemidler».
Ordlyden i legemiddelloven § 20 omfatter også næringsmidler. Næringsmidler kan ikke markedsføres med reklame som inneholder medisinske påstander. Slik reklame vil bety et brudd på legemiddelloven § 20 og følgelig være ulovlig. Dette følges opp i næringsmiddellovgivningen gjennom forskrift av 21. desember 1993 nr. 1385, som er fastsatt med hjemmel i næringsmiddelloven §§ 1 og 4. Den regulerer blant annet reklame for næringsmidler. I henhold til forskriftens § 5 tredje ledd jf. annet ledd nr. 2 er det ikke tillatt i reklame eller presentasjon av næringsmidler «å påstå eller gi inntrykk av at et næringsmiddel forebygger, leger eller lindrer sykdom, sykdomssymptomer eller smerter». Medisinske påstander rammes også her selv om de faktisk er sanne. Det kan imidlertid konstateres at denne forskriften har en snevrere formulering enn legemiddelloven § 20. Forskriftens reklamebestemmelse nedlegger ikke forbud mot at produktet anbefales som middel til å påvirke fysiologiske funksjoner hos mennesker. Dette representerer imidlertid neppe noen materiell forskjell. Legemiddelloven og næringsmiddellovens regulering av reklame med medisinske påstander går parallelt og de overlapper hverandre. Det er imidlertid legemiddelloven § 20 som får anvendelse for alt som ikke er legemidler, herunder næringsmidler, dersom det lovstridig anvendes medisinske påstander i reklame. At bestemmelsen har blitt plassert i legemiddelloven, er bevisst valgt av lovgiveren. Apotekhandelsplakatens regler om forbud mot medisinsk reklame ble opprettholdt for å hindre alminnelige kjøpmannsvarer solgt som medisinsk virksomme (jf. kapittel 4.6.2).
Lov av 16. juni 1972 nr. 47 om kontroll med markedsføring og avtalevilkår (markedsføringsloven) retter seg mot uriktig og villedende markedsføring. Her stiller loven sannhetskrav. Det vil si at dersom man beviser at en fremsatt påstand i en reklame er sann, så er reklamen ikke lovstridig. Ved brudd på markedsføringsloven er sanksjonsmidlene både administrative (herunder tvangsmulkt) og strafferettslige, jf. markedsføringsloven §§ 16 og 17. I henhold til spesialitetsprinsippet vil markedsføring av ikke-legemidler med medisinske påstander i utgangspunktet følge legemiddelloven. Dette er likevel ikke til hinder for at markedsføringsloven kan komme inn som et supplement.
4.6.3.6 Reaksjonssystemet ved overtredelse av legemiddelloven § 20
Overtredelse av legemiddelloven § 20 er sanksjonert med to sett av regler. Ulovlig reklame i legemiddelloven § 20 sin forstand kan bli møtt med reaksjoner fra forvaltningen, og den kan medføre straffeansvar. Strafferettslig forfølges overtredelse av legemiddelloven § 20 med bøter og/eller fengsel i inntil tre måneder, jf. legemiddelloven § 31. Skyldkravet er forsett eller uaktsomhet.
Sivilrettslig kan myndighetene med hjemmel i legemiddelloven § 20 annet ledd reagere ved å pålegge tilvirkeren eller annonsøren å sørge for at en godkjent beriktigelse blir sendt ut eller offentliggjort på tilsvarende måte som den ulovlige reklamen. Videre kan forvaltningen anlegge søksmål med krav om at varen forbys solgt under det navn som var brukt i den ulovlige reklamen. Lovgiverens begrunnelse for dette er behov for en effektiv håndheving av reklameforbudet i legemiddelloven § 20.
Det har i praksis vist seg at firmaer som har fått advarsler og blitt ilagt bøter for ulovlig medisinsk reklame, likevel har fortsatt med den ulovlige markedsføringen, i håp om at det økte salget de kan få ved medisinsk reklame kan oppveie boten. Preventivt antok man at bare trusselen om slike følger vil medføre at annonsørene ville vise større forsiktighet. Hertil kommer et mulig sanksjonsmiddel i legemiddelloven § 28 femte ledd. Denne bestemmelsen gir adgang til å ilegge tvangsmulkt dersom «frist for oppfyllelse av pålegg oversittes».
4.6.4 Særlig om naturlegemidler
Et naturlegemiddel er et legemiddel, i legemiddellovens forstand, til egenbehandling, i pakning tiltenkt den enkelte forbruker. Som virksomt stoff inngår oftest bestanddeler fra planteriket, sjeldnere fra dyreriket, i visse tilfeller også mikroorganismer, salter og mineraler i sin opprinnelige form og/eller i bearbeidet form.
Vurdering av naturlegemidler for registrering og utstedelse av markedsføringstillatelse foregår etter en forenklet søknadsprosess. Kravene til dokumentasjon av et naturlegemiddels kvalitet tilsvarer kravene for andre farmasøytiske spesialpreparater, mens kravene til dokumentasjon av sikkerhet og effekt er langt mindre omfattende og baserer seg kun på tradisjonell bruk i folkemedisinen. Ordningen med godkjenning av naturlegemidler ble igangsatt i april 1995. Siden har ca. ti naturlegemidler fått markedsføringstillatelse. Salg av godkjente naturlegemidler kan skje utenom apotek, jf. forskrift av 9. april 1996 nr. 367, til tross for at naturlegemidler like fullt er legemidler.
Undertiden kan det her forekomme at næringsmidler med legemiddellignende form etter denne ordningen blir godkjent som naturlegemiddel. Avgjørende for grensedragningen vil her som ellers være hvorledes produktet frembys i markedet. Utgis varen for å være medisinsk virksom, men baserer seg på tradisjonell bruk, vil ordningen med godkjenning av naturlegemidler gi en god mellomløsning.
4.6.5 Departementets vurdering og forslag til tiltak
Etter departementets vurdering er det fremdeles behov for regulering av området for medisinsk reklame. Formålet må være å hindre at alminnelige varer bevisst blir tillagt medisinsk virkning for å fremme salget. Markedet tilføres ikke sjelden reklame med medisinske påstander hvor det er store økonomiske interesser involvert. Denne form for markedsføring er ofte etisk betenkelig, blant annet ved at den retter seg mot svake grupper, for eksempel syke mennesker, og gir mange løfter som det ikke er dekning for. I mange tilfelle innebærer dette risiko for at hjelpetrengende benytter seg av slike varer fremfor å søke medisinsk behandling. De seriøse kan tape terreng i konkurranse med de useriøse, mens forbrukerne kan lide både økonomisk og velferdsmessig tap. Videre er det nødvendig å behandle alle produsenter likt. Produsenter av legemidler som har markedsføringstillatelse, må sørge for at markedsføringen er i samsvar med lovverket. Den som markedsfører et legemiddel må begrense sin markedsføring til å gjelde den informasjon som er funnet tilstrekkelig dokumentert av Statens legemiddelkontroll. Dette medfører kostnader for legemiddelprodusenten. Det ville derfor være inkonsekvent og urimelig å tillate omsetning av ikke-legemidler med udokumenterte legemiddelpåstander.
Samtidig er det kommet frem synspunkter om at bestemmelsen i legemiddelloven § 20 tolkes for strengt. Det anføres at en forståelse av bestemmelsen som ligger nær opp til bestemmelsens ordlyd, kan føre til urimelige resultater i strid med den alminnelige rettsoppfatningen. Som eksempel er det fremholdt at Statens legemiddelkontroll anser påstanden «hjelper mot sår hals/vond hals» som medisinsk. Dersom denne påstanden inngår i en reklame for en vare som ikke er legemiddel, vil reklamen være lovstridig.
Det er her reist et spørsmål om dagens forbud mot medisinsk reklame i legemiddelloven § 20 er hensiktsmessig utformet.
Dersom man velger å oppheve reklameforbudet i legemiddelloven § 20, vil et produkt med legemiddellignende beredningsform kunne anses som legemiddel etter lovens § 2 i det øyeblikk produktet utgis for å være medisinsk virksomt. Fortsetter omsetningen av et legemiddel uten markedsføringstillatelse, kan det reageres på som et tilfelle av ulovlig omsetning. Næringsmidler i tradisjonell forstand som markedsføres med medisinske påstander, vil reguleres av reglene om markedsføring av næringsmidler med medisinske påstander, gitt i merkingsforskriften av 21. desember 1993 nr. 1385. Velger man å oppheve legemiddelloven § 20, vil man kunne operere med ulike grenser for hvilke medisinske påstander man kan tillate. Det virker rimelig å ha den strengeste grensen for de legemiddellignende produktene. På grunn av likheten med legemidler vil forbrukerne lettere påvirkes til å tro at disse har medisinsk effekt. Markedsføringen av andre varer reguleres av markedsføringslovens forbud mot bruk av uriktige eller villedende fremstillinger. Det vil imidlertid oppstå noen svakheter ved et slikt system. For det første er begrepet reklame i legemiddelloven § 20 mer omfattende enn hva som kan subsumeres inn under uttrykkene «bestemt til eller utgis for», jf. definisjonen i legemiddelloven § 2. Videre kan man møte det problemet igjen at alminnelige varer som bevisst blir reklamert for med medisinske påstander, vil kunne kompensere eventuelt ilagte bøter ved at varen allerede kan ha blitt kjent som medisinsk virksom. Dette vil i realiteten resultere i at man vil opprettholde et forbud mot medisinsk reklame gjeldende bare for næringsmidler i næringsmiddelforskriften, dog med et svekket sanksjonssystem i forhold til legemiddelloven § 20.
Opphever man forbudet mot medisinsk reklame både i legemiddellovgivningen og næringsmiddellovgivningen, vil området i sitt hele bli regulert av markedsføringslovgivningen. Dette vil innebære at det for alle produkter vil bli tillatt med medisinske påstander i reklame dersom det kan bevises at påstandene er sanne. Helsemyndighetene har ingen instruksjonsmyndighet overfor Forbrukerombudet, og kan følgelig ikke bruke praktisering av reklameforbudet som et helsepolitisk virkemiddel. Helsemyndighetene vil dermed miste den overordnede kontroll med medisinske påstander fremsatt i reklame.
Beholder man legemiddelloven § 20 i nåværende form, kan man ved en kontinuerlig vurdering utnytte den dynamikk som ligger i bestemmelsen gjennom en tolkning og praksis harmonisert med utviklingen ellers i samfunnet. Etter departementets overveielser bør den skjønnsmessige vurderingen av hva som bør anses som medisinsk påstand, være rettet mot påstander om effekt mot definerte eller spesifikke sykdommer/sykdomssymptomer, og ikke generelle helsepåstander. Dette må dog gjøres under hensyntagen til en rimelig grad av forutsigbarhet på markedet. Videre er det viktig å sørge for en ensartet praksis, særlig overfor alle former for næringsmidler som anvender medisinsk reklame.
Departementet vil på denne bakgrunn foreta en opprydding i regelverket på området, for blant annet å unngå å regulere samme spørsmål i flere forskjellige lovverk. I tillegg er det nødvendig å styrke sanksjonssystemet i lovverket. Sanksjonsmidlene i legemiddelloven § 20 tredje ledd og § 31 første ledd forutsetter domstolsbehandling, som ofte kan være svært ressurskrevende. I praksis registrerer man at politianmeldelser henlegges på grunn av kapasitetsmangel. Sanksjonen i legemiddelloven § 20 annet ledd må antas å ha liten avskrekkende effekt overfor overtredere. Risikoen for at brudd på legemiddelloven § 20 skal få konsekvenser for markedsføreren er små, og dette må ansees som en av årsakene til at bruddene på bestemmelsen er mange. Med bakgrunn i dette er det behov for et mer effektivt sanksjonssystem i lovverket.
Departementet vil videre se nærmere på hvorvidt ordningen med registrering av naturlegemidler virker etter sin hensikt, og hvordan den bør forvaltes i fremtiden. Det er grunn til å tro at ordningen, slik den er i dag, har et forbedringspotensiale. Den har så langt ikke fungert tilfredsstillende med bare ca. ti godkjente naturmidler. Statens legemiddelkontroll har påpekt to nærliggende grunner. For det første har ordningen antakelig blitt oppfattet som kostbar av potensielle søkere. Det anvendes samme avgiftssats som ved registrering av farmasøytiske spesialpreparater. Videre har ordningen blitt oppfattet som snever. Det er et vilkår for godkjenningen at produktet egner seg for egenbehandling. Departementet understreker viktigheten av, og behovet for, at ordningen for godkjenning av naturlegemidler virker optimalt. Departementet vil således gå inn for en kraftig nedsettelse av dagens gebyr- og avgiftsnivå ved registrering av naturlegemidler, slik at ordningen blir mest mulig tilgjengelig for aktørene i markedet.
Sammenfatningsvis konstaterer departementet at det foreligger flere hensyn som tilsier at en regulering av medisinsk reklame fremdeles er nødvendig. Anvendelse av legemiddelloven § 20 forutsetter i dag en faglig skjønnsutøvelse for hva som skal ansees som medisinsk påstand i reklame. Departementet vil likevel vurdere å binde dette skjønnet i legemiddelloven § 20 med det formål å avgrense lovens virkeområde til å omfatte medisinske påstander om terapeutisk effekt mot nærmere definerte sykdommer med en viss alvorlighetsgrad. Departementet finner det ikke hensiktsmessig at påstander om medisinsk effekt på lettere sykdommer og sykdomssymptomer, som for eksempel «sår hals, irritert hals», blir rammet av loven dersom de anvendes informativt i markedsføring av produkter.
Departementet vil videre på et senere tidspunkt foreslå å endre legemiddelloven for å styrke og effektivisere lovens sanksjonssystem for imøtegåelse av ulovlig medisinsk reklame.
Departementet vil komme tilbake til spørsmålet om utvidelse av registreringsordningen for naturlegemidler ved behandlingen av Aarbakkeutvalgets innstilling om alternativ medisin.
5 Legemidler på blå resept
5.1 Stønadsordningene for legemidler
5.1.1 Lik rett og lik tilgjengelighet til legemidler
Ett av velferdsstatens sentrale mål er lik rett og lik tilgjengelighet til helsetjenester. For legemidlenes del innebærer dette at befolkningen må ha rimelig tilgang til nødvendige medisiner ved langvarig sykdom, uavhengig av betalingsevne, bosted og sosial status.
Forskrifter om stønad til dekning av utgifter til viktige legemidler og spesielt medisinsk utstyr av 18. april 1997 nr. 330 (den såkalte blåreseptordningen) er hjemlet både i kapittel 5 i folketrygdloven og i kapittel 6 i lov om vern mot smittsomme sykdommer. I det følgende beskrives det viktigste innholdet i folketrygdens mest sentrale stønadsordninger på legemiddelområdet.
5.1.2 Blåreseptordningen
5.1.2.1 Folketrygdloven § 5-14 og blåreseptforskriften
I folketrygdloven § 5-14 heter det at trygden yter stønad til dekning av utgifter til viktige legemidler og spesielt medisinsk utstyr og forbruksmateriell. Det er et vilkår for rett til stønad at medlemmet har behov for langvarig bruk av legemidlet, det medisinske utstyret eller forbruksmateriellet. Legemidlet, det medisinske utstyret og forbruksmateriellet må være forskrevet av lege.
Departementet gir forskrifter om stønad etter denne paragrafen, den såkalte blåreseptforskriften. Ordningen omtales gjerne som blåreseptordningen, ettersom legen skriver ut de aktuelle legemidlene på en blå reseptblankett. I det følgende omtales hovedtrekkene i forskriften.
5.1.2.2 Hovedregelen for å få refusjon (forskriftens § 1)
Utgifter til viktige legemidler refunderes ved behandling av nærmere angitte sykdommer. Hovedregelen for å få refusjon finnes i forskriftens § 1:
«Folketrygden yter stønad til dekning av utgifter til enkle preparater som inngår i de medikamentgrupper som er nevnt i forbindelse med den enkelte sykdom i listen i § 9.
Det ytes også stønad til sammensatte preparater, hvis disse bare inneholder medikamenter som er nevnt i listen i § 9 og ikke inneholder mer enn én representant for hver av de medikamentgrupper som er nevnt i samband med vedkommende sykdom.
Ved de sykdommer som er nevnt i § 9, jf. § 2, ytes det stønad bare hvis det er på det rene at sykdommen er gått inn i en langvarig fase og legen er overbevist om at det er behov for langvarig medikamentell behandling».
Det stilles krav om at sykdommen er gått inn i en langvarig fase og legen har overbevist seg om at det er behov for langvarig medikamentell behandling. Med langvarig forstås i praksis at pasienten må ha behov for legemidler i minst tre måneder i løpet av et år, enten sammenhengende eller i form av gjentatte, korte kurer for den samme sykdommen. Dette kan være særlig aktuelt ved stadig tilbakevendende infeksjoner eller ved sykdommer hvor plagene varierer over tid.
Kortvarig behandling gir ikke rett til refusjon, selv om det dreier seg om meget kostbare legemidler. Det er gjort unntak for legemidler til bruk mot en del smittsomme sykdommer.
Reseptfrie legemidler og handelsvarer refunderes som hovedregel ikke. Forskriften omfatter heller ikke homøopatpreparater, vanedannende legemidler (unntatt ved organisk hjernelidelse/epilepsi) eller legemidler som brukes diagnostisk. Legemidler gitt i sykehus eller sykehuspoliklinikk eller sykehjem/aldershjem dekkes av institusjonen.
Blå resepter etter § 9 skal skrives ut av lege, og kan ekspederes ved apotek uten omkostninger for pasienten utover fastsatt egenandel, og uten forhåndsgodkjenning fra trygdekontoret.
5.1.2.3 Sykdoms- og preparatlisten (forskriftens § 9)
De sykdommer som omfattes av blåreseptordningen, er listet opp i forskriftens § 9. Listen omfatter i dag 41 kroniske sykdommer eller sykdomsgrupper, og de preparatgrupper som er refusjonsberettiget ved den enkelte sykdom. Refusjonsberettigelse for det enkelte legemiddel avhenger av at det står oppført på en særskilt preparatliste utarbeidet av Rikstrygdeverket, og som distribueres til apotek og trygdekontor. Preparatlisten ajourføres etterhvert som nye preparater godkjennes for refusjon eller tidligere oppførte preparater går ut. Revisjon av listen skjer fire ganger i året. Som hovedregel blir det bare ført opp reseptpliktige legemidler med markedsføringstillatelse i Norge. Normalt må preparatene være godkjent av Statens legemiddelkontroll for den indikasjonen (sykdommen) de står oppført ved. Listen i § 9 kan også inneholder merknader om spesielle krav til forskrivning.
De fleste legemidler som er omfattet av forskriftens § 9 kan skrives ut av allmennpraktikere. Dersom det i forskriftenes merknadsfelt står at behandlingen «bør» være instituert av spesialist, er dette en veiledning til legen og ikke et vilkår for forskrivning. Dersom det står at behandlingen «skal» være instituert ved spesialavdeling i sykehus eller av spesialist i vedkommende disiplin, er merknaden å betrakte som et krav for at vedkommende legemiddel skal kunne refunderes av folketrygden på blå resept.
5.1.2.4 Legemidler som ikke er oppført i preparatlisten (forskriftens §2)
For legemidler ved kronisk sykdom som ikke omfattes av diagnoselisten i § 9, vil det unntaksvis kunne ytes refusjon etter § 2, som lyder slik:
«Det kan også unntaksvis ytes stønad til kostbare medikamenter som brukes til behandling av kroniske sykdommer som ikke er nevnt i §9 hvis medikamentet må brukes gjennom lang tid. Behandlingen bør da være instituert på spesialavdeling i sykehus eller spesialklinikk, eller av spesialist i vedkommende sykdom og bør fortsette under kontroll av vedkommende som har instituert behandlingen eller av en annen spesialist i vedkommende disiplin.
Stønad etter denne bestemmelsen må i hvert enkelt tilfelle godkjennes av Rikstrygdeverket eller den som Rikstrygdeverket gir fullmakt».
Bestemmelsen er fortolket slik at det må dreie seg om en sjelden sykdom eller om et behandlingsalternativ som sjelden er aktuelt. Det er også et vilkår at det dreier seg om kostbare medikamenter som må brukes gjennom lang tid.
Godtgjørelse etter § 2 må i hvert enkelt tilfelle godkjennes av Rikstrygdeverket eller den som Rikstrygdeverket gir fullmakt. Rikstrygdeverket har overlatt godkjenningsmyndigheten til trygdekontorene for nærmere bestemte legemidler ved spesifiserte sykdommer. Dette er i hovedsak alvorlige, kroniske sykdommer hvor diagnose er stilt i spesialavdeling i sykehus, og hvor behandling skjer i samråd med vedkommende spesialavdeling. For at legemidler kan godkjennes refundert etter § 2 skal det som hovedregel gjelde et legemiddel som har godkjent indikasjon for angjeldende sykdom. Det vil også kunne gis godkjennelse for legemidler som har markedsføringstillatelse under en annen indikasjon, eller tatt inn på registreringsfritak. For at utgiftene til et slikt legemiddel skal godkjennes refundert, kreves det at legemidlets effekt ved vedkommende sykdom er dokumentert.
5.1.2.5 Legemidler som ikke er oppført i preparatlisten (forskriftens §10, 2.ledd)
Rikstrygdeverket kan i medhold av § 10 annet ledd, når særlige grunner taler for det, godkjenne at det ytes godtgjørelse for utgifter til et legemiddel som ikke er oppført i preparatlisten ved en av de diagnoser som er nevnt i § 9. Bestemmelsen lyder slik:
«Rikstrygdeverket kan når særlige grunner taler for det, godkjenne at det ytes stønad til et legemiddel som ikke er oppført i preparatlisten ved en av de sykdommer som er nevnt i § 9».
Godtgjørelse for utgifter til et legemiddel etter § 10 annet ledd vil vanligvis bare bli gitt dersom det dreier seg om en livstruende eller kronisk sykdom, som har gått inn i en alvorlig fase og hvor de opplistede preparater ikke gir tilfredsstillende effekt eller har uakseptable bivirkninger. Ved refusjon for legemidler etter § 10 annet ledd vil det alltid bli stilt krav om at legemidlet er forskrevet av spesialist eller behandlingen instituert av spesialist. Det dreier seg vanligvis om et legemiddel som er tatt inn på registreringsfritak, eller preparater som er godkjent markedsført på annen indikasjon. For at utgiftene til et slikt legemiddel kan godkjennes refundert, kreves at legemidlets effekt ved vedkommende sykdom er dokumentert.
5.1.2.6 Allmennfarlige smittsomme sykdommer (forskriftens § 4)
I 1996 ble det innført en ny § 4 i blåreseptforskriften som åpnet for full stønad fra folketrygden til legemidler ved allmennfarlige smittsomme sykdommer. Paragrafen er hjemlet i lov om vern mot smittsomme sykdommer § 6-2.
Ordningen gjelder den som oppholder seg i Norge, selv om vedkommende ikke er medlem av folketrygden. Hvilke sykdommer og hvilke legemidler dette gjelder og hvilke indikasjoner som i tilfelle må være oppfylt, er listet opp i § 4. Det ytes også godtgjørelse for utgifter til antiinfektive legemidler og for bestemte vaksiner, immunoglobuliner og immunsera. Vaksinene rekvireres og utleveres fra Statens institutt for folkehelse som foretar vurderingen av om indikasjonen i det enkelte tilfelle er oppfylt.
5.1.2.7 Pasientenes egenandeler (forskriftens § 7 pkt. 2)
Legemiddelbrukere som har fått forskrevet et legemiddel på blå resept betaler en egenandel selv, jf. forskriftens § 7. Det skal kun betales en egenandel pr. resept. Det regnes som en resept dersom legen under en og samme konsultasjon skriver ut flere legemidler, eventuelt for flere sykdommer, til samme pasient.
Egenandelens størrelse fastsettes av myndighetene, og justeres i forbindelse med statsbudsjettet.
5.1.2.8 Referansepris (forskriftens § 7 pkt. 1)
For preparater som omfattes av referanseprisordningen, kan det fastsettes en referansepris, som er det maksimale beløp som folketrygden kan godtgjøre. Grunnlaget for denne ordningen er blåreseptforskriftens § 7 pkt. 1, som lyder slik:
«Dersom det finnes flere preparater innenfor de enkelte legemiddelgrupper som er nevnt i § 9, med samme generiske virkestoff og som regnes som medisinsk likeverdige, skal legen forskrive det billigste synonympreparat hvis ikke tungtveiende medisinske grunner tilsier noe annet. Ved igangværende behandling skal legen vurdere om det uten skadevirkninger kan skiftes over til billigste synonympreparat».
For preparater som inneholder samme virkestoff i samme styrke og legemiddelform, og som er funnet medisinsk likeverdige, kan Rikstrygdeverket fastsette en referansepris. Referanseprisen er det maksimale beløp som folketrygden kan dekke. Parallellimporterte preparater betraktes i denne sammenheng på samme måte som synonympreparater. Referanseprisen fastsettes med utgangspunkt i prisen på billigste preparat i gruppen. Egenandelen beregnes av referanseprisen. Dersom det forskrives et preparat som er dyrere enn referanseprisen, må pasienten betale differansen selv. Differansen blir ikke ført opp på egenandelskortet, og er således ikke med i beregningsgrunnlaget for utdeling av frikort. Unntak gjelder dersom leveringssvikt har oppstått for billigste preparat. I slike tilfeller refunderer trygden tilsvarende nest billigste preparat i gruppen.
5.1.3 Nærmere om referanseprissystemet
Det ble i 1993 innført et referanseprissystem for synonympreparater. Systemet innebærer at det er en øvre grense - tilsvarende prisen på billigste synonympreparat - for hvor mye trygden pliktmessig skal refundere for et preparat i en gitt gruppe synonyme legemidler.
Som følge av EØS-avtalen ble det adgang til parallellimport. For nærmere beskrivelse av parallellimport vises til kapittel 4.4 ovenfor. Parallellimport bidrar til at det eksisterer flere identiske preparater til forskjellig pris på det norske markedet. Det er vanskelig å se noen tungtveiende grunner til ikke å inkludere også parallellimporterte preparater i referanseprissystemet. Referanseprissystemet ble derfor med virkning fra 15. mars 1998 utvidet til også å gjelde parallellimporterte preparater og preparater som selges i konkurranse med parallellimporterte preparater. Utvidelsen ble beregnet å gi Folketrygden en innsparing på 140 millioner kroner.
Samlingen av referanseprisene definerer en «referanseprisliste» som er distribuert til leger, apotek, sykehjem, trygdekontor m.v. Dersom legen rekvirerer et preparat som koster mer enn referanseprisen, det vil si velger noe annet enn det billigste blant likeverdige preparater, må pasienten betale mellomlegget. Dette mellomlegget - kalt referansetillegget - blir ikke registrert på egenandelskortet, følgelig er det ikke med i beregningsgrunnlaget for tildeling av frikort. På denne måten oppstår en priskonkurranse mellom produsenter og importører av legemidler. Produsentene og importørene vet at dersom de prissetter sine medisiner høyt, vil pasienten ønske et annet legemiddel. Dette leder til at de over tid vil senke sine priser.
Dersom leveringssvikt hos grossist, direkteimportør eller parallellimportør oppstår, og apotekene av den grunn ikke kan utlevere det billigste alternativet, kan apoteket utlevere det nest billigste alternativet uten at referansetillegg avkreves.
Det har vist seg at leveringsgraden for parallellimporterte preparater i en del tilfeller er for lav. Departementet vil i løpet av 1999 tydeliggjøre overfor omsetningsleddene hvilke kriterier som skal være oppfylt før staten refunderer nest billigste preparat.
Utvidelsen av referanseprissystemet evalueres i disse dagene. Departementet vil komme tilbake til de viktigste funnene i evalueringen i forbindelse med revidert nasjonalbudsjett for 1999 eller forslag til statsbudsjett for år 2000.
Regjeringen har i sitt forslag til statsbudsjett for 1999 foreslått ytterligere utvidelse av referanseprissystemet. Utvidelsen er tredelt: opptak av flere legemiddelgrupper som av praktiske årsaker til nå ikke har vært omfattet av systemet, endring av kravet om lik farmasøytisk form (tablett, kapsel etc.) ved tildeling av referansepris, og inkludering av legemiddelpakninger med mellom 151 og 300 piller. Rikstrygdeverket og Statens legemiddelkontroll har samarbeidet om å finne hvilke legemiddelformer som kan betraktes som medisinsk likeverdige, med sikte på å gi disse ens referansepris, som erstatning for dagens krav om samme medisinske form. Fortsatt vil legemidler som tildeles samme referansepris inneholde samme virkestoff i samme mengde. Utvidelsen er beregnet å gi en innsparing for trygden på 75 millioner kroner pr. år.
Tiltaket vil kreve endring i blåreseptforskriften. De impliserte vil få en anledning til å tilkjennegi sitt syn på saken når utkast til revidert blåreseptforskrift blir sendt på høring. Nærmere spesifisering av hvilke legemiddelformer som kan betraktes som medisinsk likeverdige vil bli lagt ved høringsutkastet.
5.1.4 Bidragsordningen
5.1.4.1 Folketrygdloven § 5-22 og bidragsordningen
Allerede før 1953, da ordningen med pliktmessig stønad til legemidler ble innført, hadde flere trygdekontorer på frivillig basis ytt bidrag til livsviktige medisiner. Ordningen gikk ut på at trygdekassens overskudd skulle avsettes til et reservefond dersom årsregnskapet viste større inntekt enn utgift. Fondet kunne etter nærmere retningslinjer fastsatt av Rikstrygdeverket, anvendes til øyemed som står i forbindelse med sykepleien, således til ytelse av tannbehandling, medisin, opprettelse av eller støtte for sykehus, føde,- tuberkulose- eller rekonvalesenthjem og lignende samt til sykdomsforebyggende arbeid.
Bidragsordningen ble videreført som et supplement til blåreseptordningen og er nå hjemlet i § 5-22 i folketrygdloven. Ordningen innebærer at trygden kan yte bidrag til dekning av utgifter til helsetjenester når utgiftene ikke ellers dekkes etter denne loven eller andre lover. Utgifter til flere legemidler og bandasje- og forbruksmateriell kan ses under ett.
Pasienten må betale legemidlet på apoteket og søke trygdekontoret om bidrag på grunnlag av spesifiserte kvitteringer. Etter gjeldende retningslinjer ytes bidrag med 2/3 av de utgifter som overstiger 600 kroner i løpet av et kalenderår. Pasientens andel av disse legemiddelutgiftene teller ikke med under det totale beregningsgrunnlaget for tildeling av frikort. I Rikstrygdeverkets generelle retningslinjer er det presisert at bidrag ikke er noen pliktmessig ytelse, at det er et begrenset beløp som er stilt til rådighet for bidragsformål, at det er en forutsetning at bidragsrammen ikke skal overskrides, og at Rikstrygdeverket er pålagt å se til at så ikke skjer og om nødvendig tilpasse retningslinjene slik at rammen strekker til.
5.1.4.2 Unntaksvis kostnadsfrihet
For pasienter med kreft og immunsvikt ytes bidrag til full dekning av utgifter til avførende, beroligende, hostestillende, kvalmestillende, smertelindrende og søvnfremkallende legemidler.
Ordningen innebærer at midlene kan utleveres gratis fra apotek, selv om de forskrives på hvit resept. Apoteket sender regningen direkte til trygdekontoret.
5.1.5 Refusjon av egenbetaling - frikortordningen
Fra 1. april 1984 ble det innført en ordning med et felles årlig utgiftstak for egenandeler til legemidler forskrevet på blå resept, legehjelp, psykologhjelp og reiser som trygden betaler for.
Hensikten var å skjerme storforbrukere av helsetjenester mot for høye totalutgifter til egenandeler. Egenbetaling ved de tjenester som inngår i ordningen, kvitteres på et spesielt kvitteringskort utstedt av Rikstrygdeverket. Når samlet egenbetaling fra årets begynnelse når egenandeltaket, innløses kvitteringskortet i et frikort, og brukere belastes ikke egenandeler ut kalenderåret. For barn under 16 år beregnes utgiftene samlet for alle søsken og en av foreldrene. I statsbudsjettet for 1999 ble egenandeltaket foreslått satt til 1.550 kroner.
Det kan være grunn til å spørre om det vil være hensiktsmessig å tilby pasienten å kjøpe et frikort fremfor å la pasienten måtte samle på kvitteringer for deretter å innløse kvitteringskortet i et frikort. De mest åpenbare fordelene ved dette er at de lokale trygdekontorene vil få frigjort ressurser gjennom redusert kontrollvirksomhet, og at pasienten ikke trenger påse at kvittering blir påført kvitteringskort eller lide tap dersom egenandelskortet kommer på avveie. Departementet vil vurdere å gjennomføre en slik ordning.
5.1.6 Kostnadsveksten på legemiddelområdet
I tidsrommet fra og med 1990 til og med 1997 økte utgiftene til legemidler på blå resept med i gjennomsnitt 11,1 prosent årlig, målt i løpende kroner. Utgiftsveksten i bidragsordningen var i årene 1991 til 1997 på 22,5 prosent i gjennomsnitt pr. år. I 1997 var veksten i bidragsordningen på omtrent samme nivå som veksten i blåreseptordningen. Utgiftene til de to ordningene fremgår av tabell 5.1.
Tabell 5.1 Rikstrygdeverkets utgifter til legemidler på blå resept og stønad til legemidler etter bidragsordningen, 1990-1997
| Blåresept- | Bidrags- | |||
| ordningen | ordningen: | |||
| Årlig vekst | Årlig vekst | |||
| År | Mill.kr. | i prosent | Mill.kr. | i prosent |
| 1990 | 2 221 | 15,1 | 25,8 | - |
| 1991 | 2 561 | 15,3 | 30,1 | 16,7 |
| 1992 | 2 831 | 10,5 | 40,7 | 35,2 |
| 1993 | 3 035 | 7,2 | 53,6 | 31,7 |
| 1994 | 3 264 | 7,5 | 63,3 | 18,1 |
| 1995 | 3 665 | 12,3 | 78,0 | 23,2 |
| 1996 | 4 065 | 10,9 | 95,2 | 22,1 |
| 1997 | 4 476 | 10,1 | 105,5 | 10,8 |
Det offentliges andel av finansieringen av det samlede legemiddelforbruk har også økt det siste tiåret. Figur 5.1 viser fordelingen av legemiddelutgiftene mellom Rikstrygdeverket, sykehusene og private utlegg til egenandeler.
Figur 5.1 Offentlig og privat andel av legemiddelutgiftene
Av figuren kan man lese at staten de siste ti årene har tatt er større ansvar med hensyn til å begrense pasientenes utgifter til nødvendige medisiner. Det underliggende tallmaterialet viser dessuten at sykehusene har opplevd en vesentlig lavere utgiftsvekst i perioden enn både pasienter og stat. Det er nærliggende å anta at dette skyldes at sykehusene i større grad enn pasienter og stat forhandler om pris på legemidler.
5.1.7 Grundutvalgets vurdering av stønadsordningene på legemiddelområdet
Grundutvalget foretok en omfattende analyse av blåreseptordningen. Utvalget uttalte at sammenlignet med tilsvarende ordninger i andre land, må den norske refusjonsordningen beskrives som svært god.
Grundutvalget satte allikevel frem flere forslag til hvordan dagens blåreseptordning kan forbedres, og til hvordan myndighetene kan legge til rette for riktigere forskrivning og bruk av legemidler. Sentrale forslag presenteres i det følgende.
5.2 Pasienter med sjeldne diagnoser
5.2.1 Grundutvalgets forslag og høringen
Som vist tidligere åpner dagens blåreseptforskrift for at pasienter med alminnelige sykdommer kan få dekket sine utgifter både til kostbare og rimelige legemidler. Pasienter med sjeldne sykdommer får derimot kun utgiftene til kostbare legemidler dekket. Grundutvalget anså at dette representerte en forskjellsbehandling av pasienter, og foreslo at pasienter med sjeldne diagnoser bør få samme rett til å få sine utgifter dekket som pasienter med alminnelige sykdommer.
Høringsinstansene var positive til en slik justering av blåreseptordningen.
5.2.2 Departementets vurdering og forslag til tiltak
Fra et fordelingspolitisk perspektiv kan det neppe begrunnes at pasienter med alminnelige lidelser skal ha enklere tilgang til legemidler enn pasienter med sjeldne lidelser. Tvert imot kan det argumenteres med at det å ha en sjelden lidelse er en tilleggsbelastning i forhold til å ha en vanlig lidelse fordi det ofte er lite kunnskap og få tilbud om behandling.
Sosial- og helsedepartementet vil derfor endre blåreseptforskriften, slik at det ytes den samme stønad til pasienter med alminnelige og sjeldne diagnoser, uansett legemidlets pris.
5.3 Kortvarig, kostbar behandling av alvorlig sykdom
5.3.1 Grundutvalgets forslag og høringen
Kortvarig behandling dekkes normalt ikke over blåreseptordningen, såfremt det ikke dreier seg om medisiner til bruk mot allmennfarlig smittsom sykdom. Grundutvalget foreslo at folketrygden også skulle dekke utgifter til kostbare legemidler til kortvarig behandling, såfremt sykdommen var av alvorlig karakter. Forslaget var tenkt gjennomført ved å fastsette en ny bestemmelse i blåreseptforskriften med tilhørende liste over kortvarige, alvorlige sykdommer og korresponderende legemidler.
Flertallet av de høringsinstansene som har uttalt seg om forslaget, er positive til en slik utvidelse av retten til refusjon, herunder Statens helsetilsyn og LO. Rikstrygdeverket og Finansdepartementet uttrykker seg imidlertid kritisk til forslaget.
5.3.2 Departementets vurdering og forslag til tiltak
Utgifter til behandling av allmennfarlige, smittsomme sykdommer dekkes allerede i dag av folketrygden. Alvorlige, akutte sykdommer behandles som regel i sykehus, hvor behandlingen er gratis. For en del tidlig utskrevne pasienter forutsettes det imidlertid ofte behandling også etter at de har kommet hjem. For legemidler som ikke omfattes av blåreseptordningen kan det søkes trygdekontoret om dekning av 2/3 av legemiddelutgifter over 600 kroner.
Departementet legger vekt på at pasienter med kortvarige sykdommer sjelden har den samme økonomiske totalbelastning som pasienter med kroniske lidelser. Departementet anser at tilbudet til akutt syke pasienter, som trenger kostbare legemidler som ikke dekkes av blåreseptordningen, er tilstrekkelig ivaretatt gjennom øvrige stønadsordninger, og vil ikke gå inn for noen endring av praksis eller av regelverk når det gjelder refusjon av utgifter til kortvarige lidelser.
5.4 Smertestillende legemidler til spesielle grupper kronikere
5.4.1 Smertestillende legemidler med vanedannende effekt
Ved spørsmål om opptak av legemidler i blåreseptordningen har det vært et prinsipielt skille mellom legemidler som kan være vanedannende og andre legemidler. Av hensyn til misbruksproblematikken har man ikke ønsket å inkludere legemidler som kan være vanedannende. Dette gjelder smertestillende og beroligende legemidler. Erfaringene fra den gang det var anledning til å forskrive beroligende legemidler på blå resept under punkt 18 (i sykdomslisten under § 9 i blåreseptforskriften), viste at disse i stor grad ble forskrevet i strid med forskriftene og at misbruksfaren var betydelig. Det var angivelig et press på legene for å forskrive vanedannende legemidler på blå resept. Trygdekontorene og apotekene har på sin side liten eller ingen mulighet til å kontrollere om legemidlene forskrives i samsvar med reglene eller om det utvikler seg et misbruk.
Selv om en av prinsipielle grunner ikke har ønsket å inkludere smertestillende legemidler som kan være vanedannende i blåreseptordningen, betyr det ikke at en ikke har erkjent behovet for i visse situasjoner å kunne yte stønad til slike legemidler. Dette er bakgrunnen for at det etter gjeldende retningslinjer kan ytes bidrag til full godtgjørelse av utgifter til smertestillende legemidler ved sykdommer som kommer inn under blåreseptforskriften § 9, punkt 9 (cancer) og 38 (immunsvikt). Det er et vilkår at sykdommen har gått inn i en langtkommen og uhelbredelig fase og at faren for misbruk er bedømt som underordnet. I disse tilfellene er det innført en forenklet oppgjørsordning slik at apoteket kan utlevere legemidlet dersom legen har anført «§ 5-22, kfr. § 9/38» på resepten. Pasienten skal ikke betale egenandel. Det kreves heller ikke forhåndsgodkjennelse fra trygdekontoret. Denne ordningen er gunstigere for pasienten enn hva som gjelder for legemidler som er opptatt på blå resept, fordi det ikke skal betales egenandel. Pasienter med langtkommen, uhelbredelig kreft eller immunsvikt står således i en særstilling når det gjelder stønad til legemidler som kan være vanedannende. Begrunnelsen for dette er pasientenes begrensede leveutsikter, som gjør at misbruksfaren bedømmes som av underordnet betydning.
Å utvide ordningen til å omfatte alle pasienter med begrensede leveutsikter og hvor sykdommen ledsages av smerter synes i utgangspunktet å være et rimelig krav, men vil skape vanskelige avgrensningsproblemer både medisinsk og administrativt. Sosial- og helsedepartementet er derfor av den oppfatning at det eksisterende skillet bør opprettholdes, og at smertestillende legemidler som kan være vanedannende, som hovedregel ikke bør inngå i blåreseptordningen. Pasientene vil imidlertid, som andre pasienter med kroniske smerter, kunne få bidrag til delvis godtgjørelse av utgifter til smertestillende midler etter de vanlige retningslinjer, det vil si med 2/3 av utgiftene utover et minstebeløp på 600 kroner pr. kalenderår. Det er et vilkår at behandlingen er instituert av spesialist i sykehus og at behandlingen vurderes med to års mellomrom. Faren for avhengighet må være bedømt som helt underordnet.
5.4.2 Smertestillende legemidler uten vanedannende effekt
De fleste reseptpliktige legemidler som virker smertestillende, men som ikke kan gi avhengighet, dekkes i dag av blåreseptordningen.
Det gjelder eksempelvis ikke-steroide antiinflammatoriske legemidler (NSAID-preparater) som refunderes ved smertetilstander etter punkt 11 (morbus collagenus), punkt 17 (polyarthritis chronica) og punkt 35 (alvorlig symptomgivende cox- og gonartrose) i sykdomslisten under § 9 i blåreseptforskriften. Smertestillende midler mot angina pectoris refunderes etter punkt 12 (morbus cardiovascularis) og midler mot migrene etter punkt 36. Enkelte antiepileptika refunderes etter punkt 22 ved trigeminusnevralgi, som er en svært smertefull tilstand.
5.4.3 Departementets vurdering og forslag til tiltak
Ifølge Rikstrygdeverket er den mest iøynefallende mangel ved smertebehandlingstilbudet i dagens blåreseptordning at det ikke gis stønad til legemidler til bruk ved nevrogene smerter (nervesmerter), først og fremst som følge av diabeteskomplikasjoner. Dette gjelder antiepileptika (karbamazepinpreparater) og tricykliske antidepressiva. Både karbamazepinpreparater og tricykliske antidepressiva er rimelige legemidler med et relativt begrenset forbruk i befolkningen.
Sosial- og helsedepartementet anser at ovennevnte diagnosegrupper tilfredsstiller vilkårene for å kunne omfattes av blåreseptordningen, og er positiv til å utvide ordningen til den bruk som her er beskrevet. Det gjenstår imidlertid en del spørsmål knyttet til den praktiske avgrensningen av en ordning. Departementet vil komme tilbake til denne saken i 1999.
5.5 Like egenandeler
5.5.1 Grundutvalgets forslag
Alders- og uførepensjonister har lenge hatt lavere egenandel for legemidler på blå resept enn andre pasienter. Det samme gjelder barn under 16 år. Fra 1991 er barn under 7 år fritatt for egenandeler i helsetjenesten. Differensieringen av egenandelen etter alder og pensjonsstatus er en særordning for legemidler på blå resept. Heller ikke utgiftstaket for egenandeler i frikortordningen, som for 1999 er foreslått til 1.550 kroner, er differensiert etter disse kriteriene, men barnefamiliene er tilgodesett ved at barns egenandeler kan summeres sammen med den ene av foreldrenes. Alders- og uførepensjonistene har som gruppe hatt en god inntektsutvikling, og dette vil fortsette i årene framover. Både blant pensjonistene og i den øvrige befolkningen varierer inntektsforholdene mye. Formålet med å innføre prosentvis egenandel for legemidler var å stimulere til nøkternhet i forskriving og bruk av legemidler. For at ikke utlegget ved den enkelte ekspedisjon skulle kunne bli så høyt at det ville skape problemer for enkelte pasienter, er det fastsatt maksimumsbeløp pr. resept. Både dette maksimumsbeløpet og det samlede utgiftstaket for egenandeler begrenser virkeområdet for den prosentvise egenandelen.
Grundutvalget mente at frikortordningen ga meget god skjerming mot en urimelig samlet egenandelsbelastning. Utvalget så heller ikke grunn til å særbehandle pensjonister når det gjelder egenandelen på blå resept. Når det gjelder nivået på dagens egenandeler, anså imidlertid utvalget at det nåværende maksimumsbeløpet på 330 kroner var i høyeste laget, både som enkeltutlegg og sett i forhold til det daværende egenandelstaket på 1.290 kroner. Taket nås med fire slike egenandeler i løpet av et år, mens det for eksempel trengs 15 allmennlegekonsultasjoner. Lav egenandel for pensjonister o.a. på inntil 110 kroner, gir på den andre siden små insentiver til en nøktern rekvireringspraksis fra lege. På denne bakgrunn foreslo utvalget at det skulle innføres en felles egenandel på 20 eller 25 prosent, med et maksimumsbeløp pr. resept på 200-250 kroner. Samlet sett var det antatt at dette ville gi omlag samme provenyeffekt som dagens egenandeler. Det ble ikke foreslått endringer for de grupper som er helt fritatt fra egenandeler.
5.5.2 Høringsinstansenes syn
Høringsinstansene til NOU 1997:7 var svært delte i synet på forslaget om å innføre den samme prosentvise egenandel til legemidler på blå resept for alle. De høringsinstanser som er kritisk til forslaget begrunner dette særlig med at enkelte grupper av eldre fortsatt må anses som så økonomisk svake at de bør skjermes gjennom lavere egenandel. Statens helsetilsynuttrykker en betinget støtte til forslaget, og uttalte:
«Gitt at det medfører riktighet at dagens pensjonister har en økonomisk situasjon som tilsvarer situasjonen for andre befolkningsgrupper som f.eks. småbarnsfamilier, og at egenandelstaket opprettholdes på et moderat nivå, støtter Helsetilsynet utvalgets forslag om lik egenandel for alle (med unntak av barn under 7 år)».
Landsorganisasjonen (LO)mener at enkelte grupper eldre fortsatt bør ha en lavere egenandel, og uttalte:
«Når det gjelder forslaget om lik egenandel for alle over 16 år, er LO av den oppfatning at mange pensjonister fortsatt har lav inntekt. Fortsatt er om lag 40% av alderspensjonistene minstepensjonister. En kostnad på 250 kroner kan være vanskelig å bære for denne gruppen. Det forekommer også at man må løse inn flere resepter samme måned. Alders- og uførepensjonister med minstepensjon bør derfor fortsatt skjermes gjennom lavere egenandel».
5.5.3 Departementets vurdering og forslag til tiltak
Sosial- og helsedepartementet anser ikke, isolert sett, at personer i høy alder har et større behov for å få dekket sine legemiddelutgifter enn andre grupper med stort forbruk av legemidler. Hensynet til storforbrukere av legemidler vil fortsatt være ivaretatt gjennom skjermingsordningen, som sikrer alle gratis legemidler når egenandelene overskrider en viss grense.
Sosial- og helsedepartementet mener det bør være lik egenandel for alle, men anser at den bør ligge på 36 prosent, jf. St prp nr 1 for 1998-99. De grupper som i dag er fullt fritatt for egenandel, bør fortsatt være det.
5.6 Forholdet mellom legemiddelbruk i og utenfor institusjon
5.6.1 Grundutvalgets forslag
Sykehusene har ansvaret for all legemiddelbruk under sykehusopphold. Ved sykehusopphold har pasienten krav på gratis medisiner. Medisiner forskrevet til pasienten skal derfor ikke brukes under pasientens opphold i sykehus, eventuelt ved korttids- og avlastningsopphold i sykehjem.
Grundutvalget anså at dette kunne virke upraktisk, blant annet fordi det enkelte ganger må kjøpes inn ofte kostbare legemidler som ikke inngår i sykehusavdelingens ordinære legemiddelutvalg. Etter få dagers bruk vil resten av pakningene ofte måtte kasseres. Grundutvalget foreslo å myke denne praksisen noe opp. Det ble således foreslått at det skulle gis anledning til at det i sykehusavdeling, og ved korttids- og avlastningsopphold i sykehjem, kunne brukes av pasientens egen blåreseptinnkjøpte legemidler de første dagene dersom avdelingen ikke hadde det aktuelle legemiddel for hånden.
5.6.2 Høringsinstansenes syn
Bare et fåtall høringsinstanser uttalte seg om dette forslaget, og flere av disse er positive til forslaget. Det pekes imidlertid på at det kan skapes usikkerhet om hvem som har ansvaret for å fremskaffe legemidler ved korttidsopphold ved sykehus/institusjon. Landsorganisasjonen (LO)er på den annen side skeptisk til forslaget og uttaler:
«Selv om enkelte sykehus allerede har innført en praksis der pasientenes blåreseptinnkjøpte medisiner kan benyttes de første dagene, virker utvalgets forslag om å utvide dette lite gjennomtenkt. Prinsippet om gratis behandling på sykehus er viktig å opprettholde. Gjennom utvalgets forslag kan dette prinsipp lett utvannes. Resultatet av et slikt forslag kan lett bli større forskjellsbehandling og uryddige forhold knyttet til ansvaret for å fremskaffe viktige medisiner ved korttidsopphold på sykehus/institusjon».
5.6.3 Departementets vurdering og forslag til tiltak
Sosial- og helsedepartementet anser at en oppmykning av dagens ansvarsforhold med hensyn til hvem som har ansvaret for medisinforsyningen i sykehus og sykehjem vil kunne skape et regelverk som ikke er tilstrekkelig klart. Departementet deler langt på vei Landsorganisasjonens bekymring for at en endring i retning Grundutvalgets forslag, kan utvanne prinsippet om at legemidler til bruk i sykehus skal være gratis for pasienten. En slik endring kan også føre til en forskyvning av behandlingsutgifter fra sykehusene til staten via refusjonsordningen.
Departementet finner på denne bakgrunn ikke grunnlag for å endre dagens regelverk eller praksis i dette spørsmål.
5.7 Etablering av faggrupper på blåreseptordningens viktigste terapiområder
5.7.1 Grundutvalgets forslag
I NOU 1997:7 foreslo flertallet å opprette faggrupper på utvalgte terapiområder for å oppnå god forankring av refusjonsordningen i helsetjenesten. Disse gruppene skulle oversette de generelle prioriteringsprinsippene til den kliniske hverdag. For å få til dette må en klarlegge hva en skal legge i begrepene alvorlighet, effekt, kostnadseffektivitet og behandlingsintensjon for ulike diagnose- eller sykdomsgrupper, og belyse hvordan dette skal dokumenteres i behandlingen av den enkelte pasient. På bakgrunn av disse vurderingene skulle gruppene innenfor sine områder vurdere om det er behov for å revidere nåværende diagnoseliste, det vil si om noen av de indikasjonene og legemiddelgruppene som er oppført i dagens blåreseptordning ikke bør høre inn under ordningen og om forskrivningsvilkår bør presiseres. Det ble foreslått at gruppene burde representere brukerne og ha 8-10 medlemmer blant annet fra spesialisthelsetjenesten, primærhelsetjenesten, farmasi og økonomi. Gruppene bør etableres trinnvis, med hjerte- og karsykdommer som første terapiområde.
Grundutvalget foreslo også en egen blåreseptnemnd som kan fungere som et formelt samhandlingsorgan mellom forvaltningsmyndighetene og de kliniske miljøer, og legge til rette for generell kompetansebygging.
5.7.2 Høringsinstansenes syn
Et bredt flertall av de høringsinstanser som uttalte seg om dette forslaget gir sin tilslutning til slike etableringer. Legemiddelindustriforeningen (LMI)uttaler seg positivt, og mener slik etablering kan bidra til å øke kunnskapen og å heve det faglige nivået innenfor terapiområdene. Landsorganisasjonen (LO) støtter forslaget, men uttaler at en for å få en bedre test på om prioriteringskriteriene er operasjonaliserbare bør velge et vanskeligere terapiområde enn blodtrykksbehandling. Rikstrygdeverket uttaler:
«Rikstrygdeverket er enig i at det er et stort behov for at det på de ulike sykdomsområder utføres et arbeid for å bidra til at legene i sin kliniske praksis i større grad enn i dag tar hensyn til om vilkårene for forskrivning av et legemiddel på blå resept er oppfylt».
Statens helsetilsyn støtter forslaget og uttaler blant annet:
«Faggruppene bør også bistå myndighetene i den overordnede oppfølging av om prinsippene følges opp i praksis. Disse må være bredt sammensatt, tverrfaglige, ha god representasjon fra primærhelsetjenesten og også muligens ha forbrukerrepresentasjon».
Statens legemiddelkontroll støtter mindretallet i utvalget, og mener arbeidet i slike grupper i stor grad vil bli en gjentagelse av det arbeidet som allerede er utført i forbindelse med markedsføringstillatelsen. Statens legemiddelkontroll presiserer at det i forbindelse med godkjenningsprosedyren foreligger betydelig bakgrunnsdokumentasjon, samtidig som det er innhentet uttalelser fra leger med spesialistkompetanse innenfor de aktuelle sykdomsgrupper hvor legemidlet har sin effekt. Etter Statens legemiddelkontrolls mening foreligger det ikke behov for slik gjentagelse. Legemiddelkontrollen uttaler videre:
«Statens legemiddelkontroll kan ikke se at det er behov for faggrupper som premissleverandør for blåreseptordningen innenfor enkelte terapiområder i tillegg til en rådgivende blåreseptnemnd...»
5.7.3 Beskrivelse av dagens nemnder, rådgivende grupper m.v.
Allerede i dag er det en rekke rådgivende faggrupper på ulike nivåer i legemiddelforvaltningen:
Spesialitetsnemnda gir råd i spørsmål om markedsføringstillatelse for alle legemidler med nye virkestoffer. Den er bredt sammensatt med ni eksterne medlemmer og har ca. ti møter i året.
Bivirkningsnemnda gir råd i saker om sikkerhet ved bruk av legemidler. Den har seks medlemmer og møtes fire ganger i året.
Terapiverkstedene arrangeres i samarbeid med Läkemedelsverket i Sverige og organiseres som faggrupper. Det er syv verksteder i året. På hvert verksted er det 25-30 deltakere som representerer ulike deler og nivåer av helsevesenet: Sykehusleger, praktiserende spesialister, allmennpraktiserende leger, farmakologer, farmasøyter, representanter for norsk og svensk legemiddelkontroll og andre representanter. Statens legemiddelkontroll samarbeider med Den norske lægeforenings spesialistorganisasjoner for å finne frem til leger som kan delta. Hvert verksted tar for seg et sykdomsområde, og det utarbeides anbefalinger for terapi ved ulike stadier av sykdomsutviklingen. Terapianbefalingene gir et helhetlig bilde av et sykdomsområde, og legemidlenes plass i behandlingen.
Arbeidsutvalget for klinisk utprøving har fire eksterne medlemmer og har hatt åtte møter i året. Arbeidsformen er under vurdering.
Statens legemiddelkontrolls rådgivende gruppe i antibiotikaspørsmål har fem eksterne medlemmer og møtes fast to ganger i året. I perioden mellom møtene korresponderer Statens legemiddelkontroll med medlemmene om enkeltsaker.
Statens legemiddelkontrolls rådgivende gruppe for blodprodukter har fem eksterne medlemmer. Gruppen møtes ved behov.
Rådgivende gruppe for veterinærmedisin har syv eksterne medlemmer. De møte en til to ganger i året og korresponderer med Statens legemiddelkontroll om sakene.
De beskrevne nemndene fungerer etter Sosial- og helsedepartementets oppfatning bra. De er bredt sammensatt og ivaretar et helhetssyn ut fra sine funksjoner.
5.7.4 Departementets vurdering og forslag til tiltak
Sosial- og helsedepartementet anser det riktigere å bygge på de grupper og organer som er i arbeid i dag enn å bygge opp et nytt og ressurskrevende, landsdekkende nettverk som gis oppgaver som allerede langt på vei ivaretas av andre offentlige myndigheter og faglige grupperinger.
En oppbygging av et nytt nettverk vil være svært ressurskrevende, og det er usikkert hvilket gjennomslag gruppenes anbefalinger vil få på den kliniske praksis. Myndighetenes behov for å utrede begreper som alvorlighet, effekt og kostnadseffektivitet ved ulike sykdomstilstander og behandlingsalternativ dekkes dessuten i dag på en rekke områder og kan utvides etter behov.
5.8 Legemiddeløkonomiske analyser og retningslinjer
5.8.1 Grundutvalgets forslag og høringen
Grundutvalget foreslo at legemiddelprodusenter skulle begrunne sine søknader om refusjonsberettigelse for nye legemidler gjennom legemiddeløkonomiske evalueringer. Slike evalueringer går ut på at en sammenligner nytte og kostnader ved det legemidlet det søkes refusjon for, med andre aktuelle alternativer. Videre foreslo Grundutvalget at undersøkelsene skulle gjennomføres med utgangspunkt i et sett av veiledende retningslinjer, utarbeidet av myndighetene og basert på internasjonal litteratur og erfaring på feltet.
Få høringsinstanser hadde kommentarer til forslaget i høringsrunden for NOU 1997:7. De uttalelser som ble gitt, stilte seg imidlertid positive til en utvikling av fagområdet legemiddel- og helseøkonomi.
5.8.2 Retningslinjer for legemiddeløkonomiske analyser
Statens legemiddelkontroll har i ett år arbeidet med utforming av retningslinjer for utarbeidelse av legemiddeløkonomiske analyser som er tilpasset norske forhold. Retningslinjer for gjennomføring av legemiddeløkonomiske analyser må i stor grad basere seg på internasjonal metodologi og kunnskaper om legemidler og deres helsegevinster. Enkelte elementer, som for eksempel kostnadsforholdene i helsevesenet, må derimot tilpasses norske forhold for å få representative konklusjoner. Som grunnlag for dette arbeidet har Statens legemiddelkontroll benyttet tilsvarende retningslinjer fra Canada og Australia, hvor slike retningslinjer har eksistert siden begynnelsen av 1990-tallet. Tilsvarende utviklingsarbeid foregår i flere andre europeiske land. England har ferdigstilt kortfattede retningslinjer, mens flere andre land fortsatt er i ferd med å utarbeide slike. Statens legemiddelkontroll har i løpet av året fått tilgang til de engelske retningslinjene samt et utkast til danske og franske retningslinjer. Pr. i dag er det bare Australia, som har hatt retningslinjer siden 1992, som har gjort slike analyser til en obligatorisk del av legemiddelprodusentenes søknader om refusjon.
Det arbeidet som har vært utført ved Statens legemiddelkontroll har skjedd i samarbeid med en støttegruppe bestående av representanter fra legemiddelindustrien og Sosial- og helsedepartementet. Retningslinjene er sendt til uttalelse til de øvrige statlige legemiddelmyndigheter, Legemiddelindustriforeningen, Den norske lægeforening m.fl.
Over år vil det kunne utvikles kunnskap og erfaring vedrørende slike analyser. Opplysninger om økonomiske forhold vil forhåpentligvis bli registrert parallelt med innsamling av opplysninger om legemidlenes sykdomsbekjempende effekt, ideelt sett også på nasjonalt nivå.
5.8.3 Departementets vurdering og forslag til tiltak
Sosial- og helsedepartementet anser at legemiddeløkonomiske analyser vil gi myndighetene et bedre utgangspunkt for å fastlegge omfanget av blåreseptrefusjoner, gi produsentene mer stabile og oversiktlige rammevilkår og gi leger og sykehus mulighet til å treffe mer rasjonelle beslutninger.
Departementet vil understreke at kompetansen til å utføre og evaluere slike analyser er begrenset, både i Norge og internasjonalt. Det finnes heller ikke ett sett av legemiddeløkonomiske retningslinjer som er riktige i enhver sammenheng, og det er nødvendig å gjennomføre, tolke og bruke legemiddeløkonomiske analyser med varsomhet. Retningslinjer må derfor ses på som en første del av et regelverk under utvikling over tid, som vil bli forbedret av de statlige legemiddelmyndigheter i dialog med berørte parter og kompetansemiljøer. Samtidig er det ønskelig å ha et premissgrunnlag som i størst mulig grad er felles for aktørene. Ovennevnte arbeid vil bli videreført med et slikt siktemål.
Departementet vil for øvrig slutte seg til Lønning II-utvalgets syn på bruken av helseøkonomiske analyseredskaper, jf. NOU 1997:18 «Prioritering på ny»:
«Analyseredskapenes rolle er å bidra til å informere beslutningstakere. De erstatter ikke vanskelige verdibaserte valg, omhyggelig vurdering, godt klinisk skjønn eller sunn fornuft. De kan således ikke være det eneste beslutningsunderlaget for prioriteringsbeslutninger. Andre betraktninger, om likhet, rettferdig fordeling, tilgjengelighet, individuelle preferanser m.m. må også legges til grunn. Derfor er det viktig at disse analyseredskapene brukes med kunnskap om hvor langt de rekker, og at de suppleres med forståelse for hvordan sykehus og andre enheter i helsevesenet er organisert, og for det medisinske arbeidets karakter. Ikke minst er det viktig å få oppøvd evnen til etisk refleksjon».
5.9 Større offentlighet rundt spørsmål om opptak på blå resept
5.9.1 Praksis i saker om blåreseptopptak av nye legemidler
Spørsmål om nye legemidler skal kunne rekvireres på blå resept, og dermed subsidieres av folketrygden, er ofte gjenstand for stor interesse i media, blant pasienter, leger og politikere, og ikke minst hos legemiddelbedriftene. Ved at et nytt legemiddel kommer på blå resept gjøres det tilgjengelig for alle som oppfyller de medisinske kriteriene for å bruke legemidlet. Refusjonsberettigelse for nye legemidler kan i en viss grad også virke som et signal til legene om at et nytt legemiddel kan ses som en innarbeidet og akseptert del av det medikamentelle terapitilbudet. Blåreseptstatus leder derfor ofte til at forbruket av et legemiddel øker kraftig, og at folketrygdens utgifter øker tilsvarende. Spørsmål om refusjon av utgifter til nye og viktige legemidler inngår derfor i den ordinære budsjettprosessen, hvor nye legemidler må prioriteres i forhold til andre tiltak på helsesektoren.
I vurderingen av om nye legemidler skal komme på blå resept utfører Statens legemiddelkontroll analyser av kostnadene ved bruk av legemidler sammenholdt med de helsegevinster bruken gir, og eventuelle innsparinger på andre områder. Analysene utføres i hovedsak på oppdrag fra Sosial- og helsedepartementet eller Rikstrygdeverket og er utformet som saksforberedende anbefalinger til departementet vedrørende hvilken refusjonsstatus et legemiddel skal ha. Dokumentene er en del av det underlagsmateriale Sosial- og helsedepartementet benytter som grunnlag for budsjettsaker. I henhold til offentlighetsloven §§ 5 og 6 kan dokumentene unntas offentlighet.
5.9.2 Departementets vurdering og forslag til tiltak
Sosial- og helsedepartementet har som mål å skape større offentlighet rundt spørsmål om opptak av nye legemidler på blå resept. Dette målet må vurderes opp mot behovet for å unnta budsjettspørsmål fra offentlighet i utredningsfasen og legemiddelindustriens krav på konfidensiell behandling av forretningssensitivt materiale.
Etter at vedtak om refusjonsberettigelse er fattet, tar departementet sikte på at de legemiddeløkonomiske analysene skal kunne offentliggjøres. Eventuelt kan slik offentliggjøring skje etter en tidsplan som angir når dokumentene frigis for offentlig innsyn. Statens legemiddelkontroll tar også sikte på en juridisk gjennomgang av hvilke forhold som må ivaretas i forbindelse med en offentliggjøring av analyser, slik at en unngår å komme i konflikt med eventuelle sekretessehensyn av betydning for legemiddelprodusentene.
5.10 Utredning av et terapeutisk referanseprissystem
5.10.1 Grundutvalgets forslag
Grundutvalget foreslo at mulighetene for å innføre et terapeutisk referanseprissystem for ulike grupper av legemidler skulle utredes. Det ble fremholdt at terapeutiske referansepriser kunne være en metode for å få til en mer kostnadseffektiv bruk av legemidler, men at legitimiteten til et slikt system vil avhenge av kvaliteten på de medisinsk-faglige vurderinger som legges til grunn for refusjon. Det er derfor viktig at man trekker på den beste medisinsk-faglige kompetansen på de terapiområdene som velges.
Bruk av referanseprissystemer innebærer at det for nærmere definerte legemiddelgrupper beregnes en pris som utgjør en øvre grense for refusjon. Legen står fritt til å forskrive legemidler som er dyrere enn denne referanseprisen, men det overskytende beløpet, referansepristillegget, må da betales av pasienten i tillegg til egenandelen. I 1993 ble det i Norge innført en slik ordning for synonyme (generiske) preparater.
På enkelte områder kan det være mulig å gruppere legemidler i kategorier hvor legemidlene ikke er synonyme, men terapeutisk sammenlignbare. Det innebærer at pasienten skal ha sammenlignbar medisinsk nytte av alternative behandlingsopplegg. Legemidlenes virkestoffer kan derimot være forskjellige. Referanseprissystemer basert på terapeutisk sammenlignbarhet er omstridt, og det er omfattende debatt både nasjonalt og internasjonalt om konsekvensene av slike begrensninger av refusjonene.
5.10.2 Høringsinstansenes syn
Forslaget om å utrede mulighetene for å innføre et terapeutisk referanseprissystem synes å være det av Grundutvalgets forslag som blir møtt med flest kritiske kommentarer blant høringsinstansene. Enkelte interesseorganisasjoner går langt i sin kritikk av forslaget, og fremhever særlig at økonomisk ressurssvake pasienter vil kunne bli hardt rammet av innføring av et slikt system.
Statens legemiddelkontrollog Statens helsetilsynstøtter forslaget om utredning. Sistnevnte uttaler:
«Selv om Helsetilsynet mener det først og fremst bør investeres i bedre forskrivningsvaner og gode behandlingsretningslinjer, vil vi støtte at et terapeutisk referanseprissystem utredes. Helsetilsynet anser det ikke for aktuelt å innføre terapeutisk substitusjon (rett for apotek til å bytte mellom ulike virkestoffer)».
Rikstrygdeverket går imot forslaget og mener det er knyttet betydelige betenkeligheter til et slikt system. Blant annet peker de på at selv om effekten for ulike preparater kan være omtrent like god målt på større grupper pasienter, gjelder ikke dette uten videre for den enkelte pasient.
Legemiddelindustriforeningen(LMI) uttrykker sterk skepsis mot å bruke ressurser på å utrede mulighetene for å innføre et terapeutisk referanseprissystem. Terapeutiske referansepriser vil etter deres syn gi økt forbruk av mindre effektive legemidler med en dårligere bivirkningprofil. Dette kan bidra til en lengre medisinering enn strengt tatt nødvendig og med større ulemper for pasienten som resultat. Foreningen uttaler blant annet:
«En lengre medisinering enn nødvendig vil påføre folketrygden unødvendige legemiddelutgifter, øke folketrygdens utgifter til sykepenger, redusere samfunnets skatteinntekter og gi en lavere samlet verdiskapning».
Norsk Revmatikerforbund uttaler at gjennomføring av en ordning med terapeutisk referanseprissystem vil forverre situasjonen betraktelig for mange mennesker med revmatiske sykdommer. Svært mange med revmatiske sykdommer representerer en økonomisk ressurssvak gruppe, og forbundet uttaler at de ikke kan akseptere forslaget.
Universitetet i Oslo, Institutt for farmakoterapi uttaler at de finner det lite realistisk at et arbeidsutvalg skal kunne si at alt innen en gruppe legemidler er likeverdig og dermed bør ha samme refusjonspris. Instituttet foreslår i stedet at en innfører ordninger som gjør at markedskreftene kan få virke bedre enn de gjør i dag. Instituttet uttaler blant annet:
«Eksempelvis finner vi det hensiktsmessig om forebyggende medisin kunne overlates mer til det enkelte individ og dermed markedskreftene».
5.10.3 Departementets vurdering og forslag til tiltak
Et terapeutisk referanseprissystem har både positive og negative sider. På den negative siden er det blant annet fremført argumenter om at referanseprissystemer kan bidra til nye sosiale skiller, ved at det er de ressurssterke som får tilgang til de beste legemidlene om en innfører slike ordninger. Mange frykter at lege-pasientforholdet vil svekkes dersom pasienten ikke gis anledning til å få legemidler forskrevet etter vanlige prinsipper for refusjon. Argumentene fra de statlige myndigheter for utvidet bruk av referanseprissystemer, har vært at trygden ikke bør belastes utover det som er nødvendig for en gitt medisinsk effekt.
Sosial- og helsedepartementet legger opp til at statlige legemiddelmyndigheter, i samarbeid med relevante fagmiljøer, skal utrede det faglige grunnlag for og økonomiske sider ved et eventuelt terapeutisk referanseprissystem. Det finnes flere alternativer for utforming av trygdens refusjonsbestemmelser i forbindelse med et eventuelt terapeutisk referanseprissystem. Disse spørsmål vil departementet eventuelt komme tilbake til etter at de faglige og økonomiske utredninger er foretatt.
6 Tiltak for å sikre riktig bruk av legemidler
6.1 Innledning
Legemidler er et av de viktigste virkemidlene vi har i behandling av sykdom. Ofte er legemidler eneste behandlingsalternativ. Medikamentell risikoreduksjon har dessuten fått en stadig mer sentral plass i det forebyggende arbeid, for eksempel innen store terapiområder som hjerte- og karsykdommer og osteoporose (benskjørhet).
Det er viktig at legens beslutning om å bruke legemidler bygger på best mulig dokumentert og nøytral kunnskap og en helhetlig vurdering av pasientens egne ønsker og behov. Når en gjennomtenkt beslutning om forskrivning av legemiddel er tatt, er det viktig at legemidlene håndteres og brukes riktig, slik legen har ment.
Det ble i 1997 omsatt legemidler i Norge for 8,9 milliarder kroner. Av dette ble omkring 70 prosent dekket over offentlige budsjetter. De offentlige utgiftene til legemidler har de siste tiårene økt sterkere enn de øvrige utgiftene i helsevesenet. En stor del av økningen skyldes overgang til nye, dyrere men også ofte mer effektive legemidler. Kunnskapen om de nye legemidlene er ved godkjenningstidspunktet begrenset og basert på studier i utvalgte pasientgrupper.
Andelen eldre i befolkningen øker. Med økende alder følger økt sykelighet og ofte et høyt legemiddelforbruk, ikke sjelden med samtidig bruk av 10-15 legemidler. Dette kan gi bivirkninger og komplikasjoner og i blant medvirke til dødsfall. Det antas at 5-10 prosent av alle sykehusinnleggelser har sammenheng med bivirkninger og feilbruk av legemidler.
Tiltak som sikrer riktig forskrivning og bruk av legemidler kan være avgjørende for om legemidlene blir pasientens «venn» eller «fiende». I dette kapitlet presenteres forslag til tiltak som kan bidra til riktig og rasjonell forskrivning og bruk av legemidler.
6.2 Reseptbasert legemiddelstatistikk
6.2.1 Tidligere omtaler og forslag om reseptbasert legemiddelstatistikk
Det er viktig å kunne fremskaffe data om den faktiske legemiddelbruken, som blant annet bivirkninger, effekt, sykefravær, som grunnlag for å vurdere om nytten står i forhold til kostnadene, om legemidlene forskrives rasjonelt, og også om de brukes i samsvar med intensjonene i refusjonsordningen. Det er grunn til å tro at dette ikke alltid er tilfelle.
Reseptbasert legemiddelstatistikk er tidligere omtalt i St prp nr 1 for 1996-97 og St prp nr 1 for 1997-98. Behovet for reseptbasert statistikk er også tatt opp i St meld nr 16 for 1996-97 om narkotikapolitikken, og i St prp nr 58 for 1997-98 med Regjeringens handlingsplan for redusert bruk av rusmidler (1998-2000).
I NOU 1997:7 foreslo Grundutvalget iverksettingen av et utvidet system for oppfølging, tilsyn og kontroll av legemiddelforbruket. Det ble foreslått å etablere:
«... en reseptbasert legemiddelstatistikk som gir et mer detaljert bilde av forskrivingen av legemidler enn det myndighetene i dag har tilgjengelig. Det bør opprettes en sentral database for legemiddelforbruk og forskrivning, hvor statlige myndigheter kan hente ut informasjon om egenandeler, forskrivende lege, forskrivningsvolumer, endring i forskrivningsmønster, virkningen av statlige reguleringer m.v. I dag er det for vanskelig for den enkelte lege å skaffe seg systematisk informasjon og oversikt over egen forskrivning, og det blir vanskelig å få til selvevaluering og kollegabasert læring. Myndighetene og Lægeforeningen må med utgangspunkt i den reseptbaserte statistikken utvikle bevisstheten om riktig forskrivning. I den forbindelse må ivaretakelse av personvernhensyn vurderes nøye».
Strømutvalget satte frem tilsvarende forslag om bedre legemiddelstatistikk.
6.2.2 Statistikk som grunnlag for oppfølging av legemiddelforskrivning og bruk
Fra Norge og andre land vet vi at ikke all legemiddelforskrivning er rasjonell, og at det er store variasjoner i legemiddelbruk både i og mellom land. For å kunne si noe om hvordan legemidler brukes, er det behov for egnet og pålitelig statistikk. Antibiotika gitt på liberale indikasjoner og for hyppig vil for eksempel øke faren for resistensutvikling og gir dermed reduserte behandlingsmuligheter for dem som virkelig trenger antibiotika.
Det er ofte stor oppmerksomhet rundt opptak av nye legemidler i blåreseptordningen, mot sykdommer som Alzheimer, benskjørhet og multippel sklerose (MS) m.fl. Muligheten til å følge opp hvordan legemidler faktisk forskrives og brukes er imidlertid ytterst begrenset. En forutsetning for å oppnå den effekten i en pasientgruppe som kan dokumenteres i kliniske studier, er ofte at et legemiddel brukes regelmessig i lang tid, ofte flere år. Gode data er en forutsetning for å kunne studere effekter av legemiddelbruk under realistiske forhold, slik at myndigheter og behandlende lege kan følge opp om legemidler brukes på en slik måte at forventet effekt kan oppnås, eller om mange avbryter behandlingen av ulike årsaker.
At legemidler opptas i refusjonsordningen medfører ikke rett til å rekvirere legemidlet på blå resept i alle situasjoner. Likevel skjer nær 100 prosent av forskrivningen av slike legemidler på blå resept. Dersom dette for eksempel skjedde i bare 90 prosent av tilfellene (som trolig er nærmere intensjonen) ville innsparingen for Folketrygden på årsbasis være på ca. 400 millioner kroner (1997).
God statistikk er også nødvendig for tilsynet med forskrivning av narkotiske og vanedannende legemidler. Gjennom ratifisering av FNs konvensjoner om narkotika og psykotrope legemidler har Norge forpliktet seg til å arbeide systematisk for riktig bruk av legemidler som har misbrukspotensiale. Nødvendigheten av bedre statistikk som grunnlag for et styrket tilsyn med forskrivningen av vanedannende midler, jf. Regjeringens handlingsplan for redusert bruk av rusmidler (1998-2000), er nærmere beskrevet i St meld nr 16 for 1996-97 om narkotikapolitikken.
For å kunne oppnå ønskede endringer i forskrivning, må det som et minimum finnes et troverdig statistikkgrunnlag som kan avspeile virkeligheten blant annet på en måte som leger kan kjenne seg igjen i, og som kan danne grunnlag for tilbakemelding, informasjon og veiledning. Statistikk basert på grossistsalg kan avdekke variasjoner, men ikke finne forklaringer på hva som ligger til grunn for disse. Slike muligheter vil i større grad finnes i en reseptbasert statistikk.
Med rekvirentbasert reseptstatistikk med anonymiserte reseptopplysninger kan man fremskaffe kunnskap om legers rekvireringsprofil og legemiddelforbruk fordelt på aldersgrupper og kjønn. Ved hjelp av anonymiserte individopplysninger vil det i tillegg være mulig å anslå om legemidlene brukes over kort eller lang tid, samtidig bruk av andre legemidler (for eksempel hvor mange eldre som bruker mer enn fem legemidler samtidig) og utslag av egenandelssystemer. Sammenholdt med data om forekomst (prevalens) av sykdom, kan slik statistikk gi gode indikasjoner på om rekvireringen og bruken er rasjonell. Rådgivning og veiledning kan dermed bygge på bedre innsikt enn det som er mulig i dag. Gjennom tilbakemelding kan leger få innsikt i egen rekvireringspraksis, eventuelt sammenholdt med kollegers, og en anledning til å vurdere og kvalitetssikre sin rekvireringspraksis. Det er stor interesse for slike tilbud, som må utvikles i nært samarbeid med rekvirentene selv.
6.2.3 Oppfølging av legemiddelpolitikken
Både EØS-avtalen og den generelle utviklingen har bidratt til store endringer på legemiddelområdet. Legemiddelområdet blir stadig mer komplekst og dynamisk og antallet aktører øker. Regelverket for priser og avanser er endret. Det kommer stadig flere legemidler på markedet, inkludert parallellimporterte produkter. For å møte konkurransen vil legemiddelindustrien søke å påvirke rekvireringen i retning av nyutviklede og patentbeskyttede preparater. Forslaget til ny apoteklov åpner for en friere etablering av apotek og adgang til generisk og parallell substitusjon (bytte), som er ment å bidra til lavere priser uten samtidig økning i legemiddelforbruket.
For planlegging og oppfølging av faglige og økonomiske tiltak i omsetningsleddene og innenfor refusjonsordningen (effekt av introduksjon av nye legemidler, prisdannelse, prisindekser, avanser, rabatter, egenandeler, rekvireringsrestriksjoner m.v.) er dagens statistikkmateriale ikke tilstrekkelig. Materialet er basert på grossistleddet og er for lite detaljert og for upresist. Nå som det ikke lenger er faste legemiddelpriser, men faste maksimalpriser, er det i hovedsak på apotek- og reseptnivået de reelle utsalgsprisene kommer til uttrykk og kan evalueres. Det samme gjelder eventuelle gevinster av myndighetenes tiltak for bedre kostnadskontroll, som for eksempel parallellimport og økt konkurranse i apotekleddet.
6.2.4 Situasjonen i øvrige nordiske land
Alle nordiske land har tidligere hatt grossistdata som basis for legemiddelstatistikk. De nordiske land, med unntak av Norge, har etablert eller har under etablering offentlig reseptbasert statistikk.
Danmark har hatt offentlig individbasert reseptstatistikk siden 1994. Formålet har vært å skaffe til veie en fullstendig, offentlig eid legemiddelforbruksstatistikk for å styrke de sentrale helsemyndighetenes og amtenes styring og planlegging på legemiddel- og apotekområdet, herunder en prisindeks for legemidler, samt å overvåke forbruket, prisutviklingen m.v. Det satses betydelige ressurser på analyse og bruk av dataene overfor både myndigheter og helsetjenesten. Sverige har også i mange år hatt individbasert reseptstatistikk i visse områder og har nå innført krav om personnummer og legenummer på alle resepter.
I Finland registreres alle resepter som betales over refusjonsordningen (i praksis de aller fleste) med person- og legenummer, og det er nylig etablert et system med tilbakemelding til alle leger. Island har under etablering et tilsvarende system som Finland, og bruker krypterte pasientopplysninger basert på personnummer.
6.2.5 Høringsinstansenes syn
Høringsinstansene stiller seg gjennomgående positive til forslaget i NOU 1997:7 om opprettelse av en reseptbasert legemiddelstatistikk og en sentral database for legemiddelforbruk og rekvirering. En rekke høringsinstanser påpeker i denne sammenheng viktigheten av at personvernhensyn blir tilstrekkelig ivaretatt og avklart før slik opprettelse finner sted.
Statens helsetilsyn mener en nasjonal reseptbasert legemiddelstatistikk er et helt nødvendig verktøy for tilsyn og rådgivning for å bidra til riktigere legemiddelbruk både faglig og økonomisk og for å motvirke uforsvarlig rekvirering. Helsetilsynet ser det som en grunnleggende forutsetning at hensynet til personvernet er ivaretatt. Også Den norske lægeforeningstøtter forslaget. Foreningen uttaler:
«Vi støtter fullt ut etableringen av reseptbasert legemiddelstatistikk slik at det kan utvikles en bevissthet om riktig forskrivning basert på selvevaluering og kollegabasert læring. Som vi har nevnt ovenfor kan det virke hensiktsmessig at Statens helsetilsyn og Rikstrygdeverket koordinerer tilsynsvirksomheten. Reseptbasert legemiddelstatistikk er et viktig verktøy, men det må stilles strenge krav til personvern både for lege og pasient».
Datatilsynet uttaler i forbindelse med forslaget om opprettelse av en sentral database for legemiddelforbruk og rekvirering at en forutsetter at slik opprettelse kan skje uten at pasientens identitet er kjent. Tilsynet henviser til at Rogalandsforskning har utarbeidet et opplegg som vil kunne tilfredsstille både behovet for god statistikk samt personvernet til den enkelte pasient. Dersom det vil være nødvendig å opprette et sentralt register på identitet for å utarbeide statistikken, påpeker tilsynet at dette vil være et konsesjonspliktig register etter personregisterloven § 9. Det vil etter Datatilsynets oppfatning være de samme betenkeligheter knyttet til et slikt register som til alle andre sentrale helseregistre, og etter deres oppfatning vil det være behov for en forankring i loven før det kan opprettes.
6.2.6 Departementets vurdering og forslag til tiltak
Sosial- og helsedepartementet tar sikte på å etablere en mer detaljert statistikk over forskrivning og bruk av legemidler. Hovedmålet vil være å bidra til bedre kvalitet i legemiddelforskrivning og bruk, gjennom informasjon og veiledning, samt å gi myndighetene et bedre grunnlag for planlegging og styring.
En individbasert reseptstatistikk krever lovhjemmel, opprettelse av et offentlig register og nødvendige konsesjoner i tråd med ny lov om helseregistre og lov om personregistre.
Departementet legger til grunn at innhenting av forskrivningsdata skal ta hensyn til ivaretakelse av personvernet. Tilsyn, kontroll og myndighetsoppfølging kan utføres uten at pasientens identitet er kjent. Departementet vil derfor arbeide videre med en løsning basert på disse forhold. Systemet vil i oppstartsfasen bli vurdert begrenset til et representativt utvalg av resepter/indikasjoner.
6.3 Ubenyttede medisiner - begrensninger i rekvireringsvolum
6.3.1 Grundutvalgets forslag
Mye av den medisin som skrives ut på trygdens regning blir ikke benyttet. Departementet har ingen oversikt over hvor store mengder blåresept-medisiner som kasseres, men vil som illustrasjon på hvilke økonomiske konsekvenser vi her står overfor, nevne at dersom 1 prosent av den rekvirerte blåreseptmedisinen ikke blir benyttet, vil dette utgjøre en kostnad på ca. 50 millioner kroner. Dette er ikke bare kostbart. Store legemiddellagre kan også være uheldig ut fra et medisinsk synspunkt, med manglende oversikt, dårlig etterlevelse av doseringsanvisninger og muligheten for sammenblanding mellom preparater, med tilhørende risiko for legemiddelrelaterte skader og sykehusinnleggelse.
Det kan i dag tas ut legemidler for inntil tre måneders forbruk. Dersom legen ved behandlingsstart har rekvirert nok av et legemiddel til tre måneders forbruk og hele kvantumet tas ut ved oppstart av behandling, vil det bety en vesentlig kassasjon dersom pasienten likevel ikke kan bruke legemidlet, for eksempel på grunn av bivirkninger.
Såkalte startpakninger er mindre legemiddelpakninger som varer for et kortere tidsrom enn de vanligste legemiddelpakningene. Grundutvalget foreslo at legene skulle pålegges å rekvirere startpakninger ved igangsetting av langvarig behandling, og at det skulle stilles krav til at legemiddelprodusentene skulle kunne levere slike pakninger.
6.3.2 Høringsinstansenes syn
Høringsinstansene uttaler seg gjennomgående positivt til forslaget om økt bruk av startpakninger. Giftinformasjonssentralenuttaler:
«Fra Giftinformasjonssentralens synspunkt er det uheldig at det samles opp mengder av legemidler i hjemmene, fordi det forskrives større kvantum før det er sikkert om behandlingen skal fortsette. Det øker faren for forgiftninger og feilbruk.».
Den norske lægeforening støtter forslaget om krav til startpakninger, og påpeker at startpakningen må standardiseres slik at den for eksempel inneholder et gitt antall tabletter, eventuelt et gitt antall definerte døgndoser (DDD). I motsatt fall påpeker de at man vil kunne få en uoversiktlig og uhåndterlig situasjon. Legemiddelindustriforeningen (LMI)støtter også forslaget, men legger til:
«Det norske legemiddelmarkedet er lite, og markedet for startpakninger er svært lite. Det er derfor viktig at myndighetene ikke krever at legemiddelprodusenten skal levere særnorske startpakninger».
6.3.3 Departementets vurdering og forslag til tiltak
I følge opplysninger fra Statens legemiddelkontroll er det ikke noen entydig definisjon av begrepet startpakning. Det er på markedet i dag kun tre ulike preparater som hver har registrert en tilhørende startpakning. Et av formålene som ligger bak forslaget fra Grundutvalget er at legen skal få mulighet til å forsikre seg om at pasienten tåler legemidlet og også har nytte av det, før det skrives ut et større kvantum. Hvor stor pakningen må være for å få avklart dette, vil variere. Det kan dreie seg om noen få dager dersom pasienten er allergisk mot legemidlet, noe som er relativt sjeldent. For en del legemidler vil effekten kunne avklares i løpet av en til to uker, mens det for andre vil være nødvendig med opp mot en måneds forbruk for å få avklart eventuelle bivirkninger og manglende eller utilfredsstillende effekt. Å etablere spesialtilpassede startpakninger for de ulike legemidler vil bli for omfattende og uoversiktlig, slik det er påpekt av Legeforeningen i høringsuttalelsen. Det vil også kunne innebære en merutgift for industrien som vil måtte tas igjen i prisen på startpakninger eller andre pakningsstørrelser av legemidlet. Innsparingen i legemiddelutgiftene gjennom redusert kassasjon ved bruk av særlige startpakninger vil således kunne bli svært begrenset.
Den mest solgte pakningsstørrelsen for legemidler tilsvarer tre måneders forbruk. De fleste legemidler kan også leveres i pakninger som varer i ca. en måned. For de fleste legemidlers vedkommende vil dette være tilstrekkelig lenge til at en får avklart effekt og eventuelle bivirkninger. Sosial- og helsedepartementet vil utarbeide en forskrift med sikte på at det ved oppstart av behandling med nytt legemiddel kun skal rekvireres et kvantum tilsvarende en måneds forbruk. Det vil fortsatt være mulighet til å reiterere resepten for inntil ett års forbruk (få levert ut medisiner flere ganger ved bruk av samme resept). Endringen vil således ikke medføre økt behov for legebesøk for å få fornyet resepten. Dette gjør det nødvendig å håndheve bestemmelsen om at legen skal angi doseringen på resepten, noe som i stor utstrekning unnlates i dag.
Samarbeidskomiteen for legemiddelinformasjon, SAMKOM, gjennomførte i 1994 et prosjekt for å kartlegge årsakene til at blåreseptmedisin ikke blir brukt, basert på returmedisin levert til apotek. Ifølge undersøkelsen er dødsfall den hyppigste årsaken til at medisin blir returnert (29 prosent). En tredjedel av de returnerte pakningene var uåpnede, og en tredjedel hadde mer enn halvparten av innholdet igjen. Opphør av bruken av et bestemt legemiddel kan skyldes flere forskjellige forhold, herunder bivirkninger eller manglende effekt ved oppstart, helbredelse, overgang til et annet legemiddel, innleggelse i helseinstitusjon eller dødsfall m.v. Enhver endring i medikasjonen av et legemiddel som har vært forskrevet og levert ut fra apotek, før det er brukt opp, fører til kassasjon. Det er grunn til å tro at kassasjon av legemidler skyldes endringer som inntrer etter at medikasjonen har stabilisert seg, i like stor eller større utstrekning enn ved oppstart av bruken av et legemiddel.
I følge § 5 i blåreseptforskriften må den enkelte resept ikke lyde på mer enn tre måneders forbruk av medikamenter, men kan reitereres for inntil et års forbruk. Apoteket må dog ikke utlevere medikamenter som nevnt for mer enn tre måneders forbruk om gangen. Den altoverveiende del av de legemidler som utleveres på blå resept gjelder tre måneders forbruk. En nedkorting av forbruksperioden på tre måneder, vil føre til redusert kassasjon fordi det til enhver tid vil være mindre utleverte og ubrukte mengder legemidler igjen, når det skjer endringer i medikasjonen.
Dersom det eventuelt skal leveres ut et mindre maksimumskvantum legemidler enn i dag, kan enkelte pasienter komme til å oppfatte det som en belastning fordi de må gå oftere på apoteket. Også for apotekene kan dette oppfattes som en merbelastning. Hvor belastende det vil oppfattes av pasienter og apotek å endre utleveringsbestemmelsene fra tre til for eksempel to måneder er vanskelig å vurdere. Det er viktig å se til at ikke kronikere blir rammet av eventuelle utleveringsrestriksjoner. Disse forhold taler for at en eventuell endring i første omgang bør prøves ut ved utvalgte apotek, før det tas endelig stilling til forslaget.
6.4 Dosedispensering
6.4.1 Grundutvalgets forslag og høringen
Dosedispensering innebærer at pasienten får utlevert nøyaktig det antall tabletter som vedkommende trenger, for et par ukers forbruk med den daglige dosen i hvert sitt rom i en doseringseske eller annen egnet emballasje. Dispensering kan både skje maskinelt og manuelt, og egner seg særlig godt for pasienter som har problemer med å holde styr på når de skal bruke sine legemidler og hvor mye. Det har hittil vært brukt i en viss utstrekning i hjemmesykepleien.
Grundutvalget foreslo at refusjonsordningen må bidra til dekning av utgifter til dosedispensering. Dosedispensering vil kunne gi besparelser i første rekke gjennom redusert svinn og kassasjon, og ved å lette administreringen av legemidler for pasientene. Den største gevinsten vil imidlertid være på behandlingskvaliteten. Sjansen for at legemidlene blir brukt til riktig tid og i riktig dose vil øke, særlig for pasienter som tar mange legemidler, ofte flere ganger daglig.
Forslaget om at refusjonsordningen må bidra til dekning av utgifter til dosedispensering får sterk støtte fra de høringsinstanser som har uttalt seg.
6.4.2 Departementets vurdering og forslag til tiltak
Departementet anser at kostnadene til dosedispensering vil bli begrenset, og at bruk av dosedispensering totalt sett vil gi innsparing for folketrygden gjennom mindre kassasjon av legemidler på blå resept. Det vil også kunne bidra til mindre svinn og opphoping av legemidler i private medisinforråd. Hjemmesykepleien vil få mindre belastning på personalet og økt sikkerhet i legemiddelhåndteringen, noe som kan føre til færre innleggelser i sykehus på grunn av legemiddelforgiftninger og feilbruk av legemidler. For samfunnet vil en reduksjon av ubrukte legemidler innebære økonomiske og miljømessige fordeler.
På denne bakgrunn foreslår Sosial- og helsedepartementet at folketrygden yter delvis stønad til utgifter til dosedispensering. Myndighetene vil utrede videre hvordan kostnadene til dosedispensering kan finansieres gjennom en kombinasjon av offentlige takster og fastsettelse av egne maksimale utsalgspriser på legemidler hvor dosedispensering benyttes.
6.5 Edb-baserte sykdomshåndteringsmoduler
6.5.1 Grundutvalgets forslag
Edb-baserte sykdomshåndteringsmoduler er kliniske beslutningsstøttesystemer som leder legen gjennom en diagnostisk sjekkliste som kan ende opp med en terapianbefaling. Systemene kan også kobles til pasientsystemer, og kan blant annet gi legen mulighet til å skaffe seg oversikt over hvilke legemidler som er rekvirert til ulike pasienter og når det er på tide å fornye resepter.
Salgsideen bak systemene er å bedre legenes mulighet til faglig oppdatering, og gi påminnelser og råd om rekvirering til pasienter i de aktuelle sykdomskategorier. Systemene utvikles på bestilling av legemiddelprodusentene, og en rekke systemer er i bruk i dag. Til tross for den overbevisende logikken ved tilnærmingen til systemene, må de også anses som markedsføringstiltak som skal bidra til at selskapets legemidler blir brukt fremfor generiske preparater, parallellimporterte legemidler eller konkurrentenes produkter. En annen konsekvens kan være økt forskrivning av produsentens egne legemidler.
Grundutvalget mente at myndighetene i langt sterkere grad må ta ansvar for å utvikle slik informasjon, gjerne i samarbeid med legemiddelindustrien. Dette arbeidet må ta utgangspunkt i det arbeid Statens legemiddelkontroll i dag gjør med terapiverksteder, og den erfaring Norges forskningsråd har med bruken av konsensuskonferanser for å påvirke klinikernes beslutninger. Det ble også presisert at systemene ikke utelukkende måtte gi medikamentell behandling som svar på et hvert helseproblem.
6.5.2 Høringsinstansenes syn
En del høringsinstanser stiller seg positive til forslaget om at myndighetene i sterkere grad må ta ansvar for å utvikle kliniske beslutningsstøttesystemer. Legemiddelindustriforeningen (LMI) uttaler blant annet:
«Det er positivt at man ser hvilken informasjon som allerede finnes i legemiddelindustrien innenfor dette området og at man ønsker å bygge videre på dette i et samarbeid med legemiddelindustrien».
Folkehelsastiller seg på sin side tvilende til effekten av slike tiltak, og uttaler:
«...utvalgets forslag vil innebære en ukjent mengde bortkastede ressurser fordi man setter i gang med tiltak som noen mener er viktige. Det finnes nå en betydelig internasjonal forskning om hvordan man kan påvirke profesjonell adferd, og effektive tiltak på dette området krever en helt annen gjennomtenkning av hva man ønsker å oppnå og gjennomarbeiding av hvordan man så skal greie å få det til, enn det utvalget har gjort».
6.5.3 Departementets vurdering og forslag til tiltak
Utvikling av ovennevnte systemer vil kreve kompetanse som myndighetene alene ikke rår over på de enkelte medisinske felt. Myndighetene har heller ingen erfaring med å utvikle, markedsføre og vedlikeholde slike systemer. Det er ikke gjort kostnadsberegninger for et slikt arbeid, men det må antas at timeprisen for medisinsk kompetanse og nødvendig programmeringskompetanse er høy. Dersom staten selv skal lage edb-baserte sykdomshåndteringssystemer, vil det kreve et omfattende faglig arbeid. Selv om myndighetene skulle lykkes i dette arbeidet, finnes det ingen garanti for at systemene blir tatt i bruk. Det er en kjensgjerning at legemiddelindustrien råder over langt større ressurser, og har stor gjennomslagskraft når det gjelder markedsføring av egne produkter. Nytteverdien av slike tiltak kan derfor synes svært usikker, jf. høringsuttalelse fra Folkehelsa. På denne bakgrunn vil ikke Sosial- og helsedepartementet ta initiativ til å få utviklet edb-baserte sykdomshåndteringssystemer i statlig regi.
I den grad det offentlige skal føre noen form for kontroll med edb-baserte sykdomshåndteringssystemer, utover de forhold som ivaretas direkte av reklameforskriften for legemidler, mener departementet at det i første omgang kan være ønskelig at produsentene pålegges å oppgi hvorvidt systemene er komplette i forhold til de legemidler som omsettes på det norske markedet. Det vil kunne betraktes som en svakhet ved enkelte slike systemer at de ikke inneholder en fullstendig oversikt, og brukeren bør gjøres oppmerksom på dette ved bruk av systemene. Systemene må heller ikke oppmuntre til forskrivning av legemidler på blå resept for tilstander som ikke er forutsatt dekket av denne ordningen. Departementet er også skeptisk til sammenkoplingen av slike systemer med legenes pasientjournaler.
Sosial- og helsedepartementet ser det allikevel som viktig at den delen av legemiddelinformasjonen fra legemiddelindustri overfor leger som først og fremst bærer preg av kunnskapsformidling, ikke hindres. Samtidig mener departementet det er behov for nærmere regler for hvordan markedsføring fra industri og leger skal foregå, også når det gjelder utvikling og bruk av edb-baserte sykdomshåndteringsmoduler. Sosial- og helsedepartementet vil invitere Legemiddelindustriforeningen, Legeforeningen og representanter for arbeidsgiversiden til å vurdere om det eksisterende regelverk for reklame for legemidler ivaretar etiske forhold rundt markedsføringen av legemidler, jf. påfølgende kapittel 7 om habilitetsspørsmål i legemiddelmarkedet. Her vil en blant annet kunne drøfte retningslinjer for innholdet av edb-baserte sykdomshåndteringsmoduler. Slike retningslinjer kan vurderes tatt inn i det regelverket som ligger til grunn for arbeidet i Rådet for vurdering av legemiddelinformasjon.
Dersom et slikt virkemiddel ikke fører til de ønskede resultater, vil departementet vurdere rettslige virkemidler. Etter omstendighetene vil edb-baserte sykdomshåndteringsmoduler helt eller delvis kunne omfattes av legemiddellovens reklamebegrep, jf. legemiddelloven kapittel VII om reklame for legemidler m.v. Nærmere regulering kan i så fall skje i forskrifter gitt med hjemmel i legemiddelloven § 19 annet ledd. Hvis regulering via legemiddellovens kapittel VII ikke gir tilstrekkelige resultater, må departementet vurdere å be om lovhjemmel til å forby edb-baserte sykdomshåndteringsmoduler som ikke holder seg innenfor de rammer det er grunn til å trekke for denne type legemiddelinformasjon m.v.
7 Habilitetsspørsmål i legemiddelmarkedet
7.1 Innledning
Offentlig ansatt helsepersonell, i hovedsak leger, er ofte i kontakt med legemiddelindustrien i forbindelse med deltakelse på kurs og lignende arrangementer arrangert av legemiddelindustrien.
Sosial- og helsedepartementet ser det som viktig at ren faglig aktivitet ikke hindres. Det er imidlertid en forutsetning at arrangementene ikke bringer helsepersonellet inn i habilitetskonflikter, og at det ikke skapes gråsoner hvor det blir konflikt mellom interessene til den ansatte, arbeidsgiver og pasient.
Slik konflikt kan tenkes å oppstå av flere årsaker. Det kan være vanskelig å avgjøre hvilke av legemiddelindustriens tiltak som er nøktern og nødvendig informasjon eller forskning, og hvilke tiltak som er ren markedsføring.
Legemiddelindustrien annonserer hyppig i medisinske tidsskrifter. I tillegg kan legemiddelindustrien gi legene tilbud om etterutdanning og finansiere deres forskningsprosjekter. Disse forhold kan tenkes å påvirke legene til å ta beslutninger om valg av legemidler som de ellers ikke ville ha gjort og som kanskje heller ikke er det beste valget av behandlingsform sett i et samfunnsøkonomisk perspektiv.
Legemiddelindustrien benytter betydelige midler til markedsføring og informasjon. Amerikanske legemiddelfirmaers utgifter til markedsføring utgjorde i 1993 hele 13.000 dollar pr. lege (Hodges, 1995). Med en dollarkurs på 7,50 kroner tilsvarer dette nærmere 100.000 kroner. Tyske tall viser at markedsføring utgjør mellom 15 og 20 prosent av selskapenes totale utgifter (Klepper, 1995). For de 25 største legemiddelfirmaene i USA representerte utgiftene til markedsføring 20,2 prosent av totalkostnadene i perioden 1982-86, mens andelen som gikk til forskning og utviklingsvirksomhet var 19,6 prosent (Mangolis, 1991).
7.2 Statens legemiddelkontroll
Ansatte ved Statens legemiddelkontroll arbeider med blant annet registrering og markedsføringstillatelser for nye legemidler. Statens legemiddelkontrolls avgjørelser kan ha stor betydning for bedriftenes suksess i markedet og lanseringen av nye produkter. Etatens arbeid må bygge på produsentnøytrale, faglige vurderinger. De ansatte kan derfor ikke påta seg arbeidsoppdrag for legemiddelindustrien som er i industriens interesse i det tidsrom de er ansatt ved Statens legemiddelkontroll. Dette følger av de vanlige krav til habilitet og til arbeidslivets krav om lojalitet overfor arbeidsgiver.
I forbindelse med sine arbeidsoppdrag for Statens legemiddelkontroll er flere av de ansatte i jevnlig kontakt med legemiddelprodusenter og representanter for disse. Slik kontakt er en forutsetning for arbeidet ved Statens legemiddelkontroll. Fagpersonale, slik som leger, farmasøyter, ingeniører o.a., kan representere Statens legemiddelkontroll ved kurs eller andre arrangementer eller delta i kurs arrangert av legemiddelindustrien. En forutsetning er at deltakelsen er klarert med overordnet og at deltakeren selv, eventuelt Statens legemiddelkontroll, betaler de utgifter som er forbundet med deltakelsen.
7.3 Regler for samhandling mellom legemiddelindustri og leger
7.3.1 Kontakt mellom offentlig ansatt helsepersonell og legemiddelindustri
Det er en omfattende kontakt mellom legemiddelindustrien og offentlig ansatt helsepersonell, både i og utenfor sykehus. Kontakten dreier seg om informasjon, kurs, konferanser, forskningsprosjekter m.v. og er som regel knyttet til de legemidlene det enkelte legemiddelselskap markedsfører. Legemiddelindustrien er avhengig av leger i forbindelse med forskning og ved klinisk utprøving av nye og forbedrede legemidler. Leger har på sin side behov for informasjon om eksisterende produkter fra legemiddelindustrien. Tilknytningen mellom leger og legemiddelindustrien er i dette henseende en forutsetning for forbedring og utvikling av legemidler og tilgjengelige behandlingstilbud.
Dette innebærer at legene får informasjon om nye legemidler, og informasjonen representerer således en del av legenes faglige oppdatering. Samtidig har aktivitetene til hensikt å virke salgsfremmende for firmaenes egne produkter. Faglige arrangementer for leger som arrangeres av legemiddelindustrien kan derfor innebære både kunnskapsformidling og markedsføring.
7.3.2 Gjeldende regelverk
Forskrift om reklame for legemidler av 25. august 1994 nr. 827 definerer reklame som enhver form for skriftlig og muntlig omtale, bilde, samt utdeling av gratisprøver av legemidler for mennesker og dyr, samt naturlegemidler, som er utformet i den hensikt å fremme salget eller bruken/anvendelsen.
Den 1. oktober 1994 inngikk Statens legemiddelkontroll en avtale med Legemiddelindustriforeningen (LMI)om at et råd for vurdering av legemiddelinformasjon løpende skal vurdere om legemiddelreklamen er i samsvar med reklameforskriften. Rådet arbeider uavhengig av Legemiddelindustriforeningen (LMI), og det er stilt opp nærmere regler for legemiddelinformasjon.
I tillegg er det utarbeidet veiledende retningslinjer for samarbeid mellom legestand og farmasøytisk industri, inngått for to år mellom legemiddelindustriens tidligere bransjeforeninger NOMI og No-Re-Farm (nåværende Legemiddelindustriforeningen) i 1990.
Departementet er kjent med at Legemiddelindustriforeningen (LMI) arbeider med et strengere regelverk, og prosedyrer for å utarbeide bedre etiske retningslinjer for forholdet mellom leger og legemiddelindustri.
7.3.3 Departementets vurdering og forslag til tiltak
Sosial- og helsedepartementet fremmet i Ot prp nr 13 for 1998-99 forslag til lov om helsepersonell. Det vises til proposisjonens kapittel 8 for nærmere redegjørelse for departementets forslag til regulering av problemstillinger knyttet til utilbørlig påvirkning av helsepersonell. Av forslaget til helsepersonellov fremgår det blant annet at innholdet i utilbørlighetskravet når det gjelder ekstern sponsing av forskning, etterutdanning m.v. kan reguleres nærmere i forskrift. Der kan det blant annet stilles krav til faglig forsvarlighet, faglig og produsentuavhengig innhold og vilkår for finansiering.
Sosial- og helsedepartementet ser det som viktig at den delen av legemiddelinformasjonen fra legemiddelindustri overfor helsepersonell som innebærer ren kunnskapsformidling, ikke hindres. Samtidig ønsker departementet mest mulig klare etiske kjøreregler for hvordan markedsføring fra industri overfor leger skal foregå. Det er ønskelig at økonomisk samarbeid mellom industrien og helsetjenesten finner sted på institusjonsplan. Sosial- og helsedepartementet vil invitere Legemiddelindustriforeningen (LMI), Legeforeningen, relevante legemiddelmyndigheter og representanter for arbeidsgiversiden til å vurdere om det eksisterende regelverk ivaretar hensynet til nødvendig kunnskapsformidling om legemidler, samtidig som det ivaretar etiske krav til markedsføringen av legemidler overfor helsepersonell.
7.4 Industrisponset forskningsvirksomhet ved offentlige helseinstitusjoner
7.4.1 Bakgrunn og behov
Klinisk legemiddelutprøving er en viktig del av den medisinske og farmakoterapeutiske forskning. Slik forskning kan bidra til sterkere norske fagmiljøer og er av stor betydning for så vel nasjonal som internasjonal legemiddelutvikling.
I henhold til veiledende retningslinjer for samarbeid mellom legestand og farmasøytisk industri skal forskning som foregår i legens vanlige arbeidstid (tjenesteplan) ikke honoreres.
7.4.2 Departementets vurdering og forslag til tiltak
I forbindelse med departementets arbeid med å styrke ledelse og organisasjonsutvikling i sykehus bør det tas sikte på at forskningsmidler og prosjektmidler går til den enkelte sykehusavdeling eller sykehus, i stedet for til den enkelte lege. Bare på denne måten kan avdelingsoverlegen og sykehusets øverste ledelse få innsyn i og oversikt over hvilken vitenskapelig aktivitet legene har i samarbeid med den farmasøytiske industrien. Ved enkelte sykehus har man allerede etablert ordninger for dette, for eksempel ved Rikshospitalet, Ullevål sykehus i Oslo og Radiumhospitalet. Sosial- og helsedepartementet vil understreke sykehusenes og eiers eget ansvar for å utvikle hensiktsmessige ordninger internt i eget sykehus, skaffe seg oversikt over ressursbruken og styre og lede virksomheten.
Sosial- og helsedepartementet vil be de berørte parter om å foreta en gjennomgang av gjeldende regelverk og retningslinjer for samarbeidet mellom leger og legemiddelindustrien. En vil vurdere behovet for å styrke regelverket, blant annet med sikte på at legemiddelprodusenters bidrag til offentlig ansatte legers forskningsvirksomhet, så lenge forskningen skjer i arbeidsgivers lokaler og i legenes vanlige arbeidstid (tjenesteplan), bør tilfalle institusjonens eller regionens forskningsorgan og eventuelt øremerkes det enkelte prosjekt.
8 Den statlige organisering av legemiddelforvaltningen
8.1 Ansvarsfordelingen i den statlige legemiddelforvaltningen
8.1.1 Någjeldende myndighets- og ansvarsfordeling
8.1.1.1 Innledning
Legemiddelsektoren er en sammensatt sektor i rask utvikling, som tilbyr samfunnet stadig nye behandlingstilbud. Private aktører står i all hovedsak for utvikling, produksjon og distribusjon av de produkter industrien skaper. Samtidig er legemidler en svært viktig innsatsfaktor i helsevesenet. Kostnadene til legemidler blir derfor i det vesentlige dekket av det offentlige. Internasjonalt blir omsetning av legemidler i stor grad regulert etter det mønster som gjelder for ordinære handelsvarer (konkurransepolitikk). For myndighetene er det viktig at både eksisterende og nye legemidler blir brukt på en riktig måte i helsesektoren, både helsefaglig og økonomisk. Myndighetene må derfor ha et velfungerende og «slagkraftig» forvaltningsapparat på legemiddelfeltet.
Legemiddelfeltet er en av de mest gjennomregulerte sektorer i Norge. Forvaltningsansvaret for legemidler er lagt til statlig nivå. Statsforvaltningen har historisk sett disponert et bredt spekter av virkemidler for å regulere hvilke produkter som tillates solgt, for å holde kontroll med legemiddelpriser og det offentliges utgifter og for å sikre kvalitet i produksjon og distribusjon, samt sørge for god tilgjengelighet og riktig forskrivning og bruk.
Nedenfor gjennomgås hovedtrekkene i dagens ansvarsfordeling i den statlige legemiddelforvaltning.
8.1.1.2 Statens legemiddelkontroll
Statens legemiddelkontroll har ansvaret for registrering av nye legemidler. Det innebærer at etaten foretar en faglig vurdering av nye legemidlers effekt, sikkerhet og kvalitet før produktet eventuelt registreres. I tillegg skal etaten blant annet godkjenne preparatomtale, pakningsvedlegg, pris m.v. før produktene tillates markedsført i Norge.
Etaten har også til oppgave å bidra til riktig bruk av legemidler, blant annet gjennom å presentere produsentnøytral informasjon om godkjente legemidler, og å motta og følge opp meldinger om bivirkninger. Legemiddelkontrollen har i tillegg en egen avdeling som utfører helseøkonomiske analyser av nye legemidler, herunder vurderer om prisen på nye legemidler står i rimelig forhold til den medisinske effekten, og hva samfunnet bør betale for produktet dersom det skal tilstås refusjonsberettigelse.
8.1.1.3 Statens helsetilsyn
Statens helsetilsyn har i dag et generelt tilsynsansvar for hele helsesektoren. På legemiddelfeltet innebærer det tilsyn med både de ulike leddene i legemiddelomsetningskjeden og med legers forskrivning av legemidler.
Statens helsetilsyn utsteder også konsesjoner for produksjon og handel med legemidler. Etaten har i tillegg en rekke funksjoner knyttet til apotekvesenets økonomi, og håndterer flere sentrale, faglige funksjoner på legemiddelområdet.
8.1.1.4 Rikstrygdeverket
Rikstrygdeverket har hovedansvaret for å forvalte folketrygdens utgifter til legemidler gjennom blåreseptordningen og bidragsordningen. Blåreseptordningen er den klart viktigste stønadsordningen på legemiddelfeltet, jf. kapittel 5 ovenfor. Trygdens utgifter utgjør ca. 5 milliarder kroner på årsbasis. Den årlige utgiftsveksten har i en årrekke vært sterk som følge av utvikling av nye, dyrere legemidler. I årene fra og med 1990 til og med 1997 var den gjennomsnittlige nominelle veksten i Rikstrygdeverkets utbetalinger til legemidler på blå resept på 11,1 prosent i året.
Blåreseptordningens utforming medfører to ulike administrative prinsipper for utbetaling av stønad til legemidler. Hovedregelen er at trygden yter pliktmessig refusjon uten nærmere søknad fra pasienten. Dette gjelder for vanlige sykdommer som er opplistet i blåreseptforskriftens § 9. Unntaksordninger går ut på at pasienten, eller pasientens lege, må søke trygdeetaten om refusjon i hvert enkelt tilfelle. Dette gjelder for sjeldne lidelser eller for behandlingsalternativer som sjeldent er aktuelle. Bidragsordningen gjelder for legemidler hvor det ikke ytes stønad etter vilkårene i blåreseptordningen. Pasienten må søke trygdekontoret om å få stønad, og egenandelen er noe høyere enn ved blåreseptordningen.
Ved behandling av søknader om stønadsberettigelse for nye legemidler må Rikstrygdeverket derfor blant annet ta stilling til hvilken av refusjonsordningene som er relevant å bruke. Landets trygdekontorer behandler søknader om refusjon i henhold til unntaksbestemmelsene, etter nærmere retningslinjer gitt fra Rikstrygdeverket sentralt. Etaten har i dag ansvaret for å gi Sosial- og helsedepartementet en samlet tilråding om det bør åpnes for refusjon for nye legemidler.
8.1.1.5 Sosial- og helsedepartementet
Sosial- og helsedepartementet har det overordnede ansvaret for legemiddelpolitikken. Viktige oppgaver er å utforme rammevilkår og håndtere de budsjettmessige og juridiske aspekter på feltet. Departementet er også klageinstans på en rekke saksområder.
De statlige legemiddelmyndigheter ivaretar arbeidet i ulike internasjonale fagkomiteer og fagorganer på sine respektive ansvarsområder.
8.1.2 Grund- og Strømutvalgenes forslag
Strømutvalget foreslo i NOU 1997:6 å utrede behovet for å samordne den legemiddeløkonomiske og legemiddelfaglige kompetanse. Begrunnelsen var at det i et lite land som Norge kunne være uhensiktsmessig å bygge opp denne kompetansen på flere steder. Ved å samle disse arbeidsoppgavene under en ledelse, ville det bli lettere for de ulike aktørene i markedet å forholde seg til sentralmyndighetenes behandling av legemiddelsaker.
I Grundutvalgets rapport NOU 1997:7 ble spørsmål om innhold i og organisering av arbeidet knyttet til blåreseptordningen viet relativt stor oppmerksomhet. Det ble presentert to hovedalternativer for organisering av arbeidet, dog uten at utvalget la frem tilrådinger om valg av modell. I den ene modellen ble Rikstrygdeverket tillagt et overordnet ansvar for behandling av refusjonssøknader, mens Statens legemiddelkontroll fikk hovedansvaret i den andre modellen. Det ble pekt på at det uavhengig av modell var behov for tett samarbeid mellom de statlige fagetatene, og at ansvaret for saksforberedelsen i refusjonsspørsmål måtte klargjøres.
8.1.3 Departementets vurdering og forslag til tiltak
8.1.3.1 Departementets vurdering
Etter Sosial- og helsedepartementets vurdering eksisterer det et visst forbedringspotensiale knyttet til håndtering av blåreseptordningen og utførelse av funksjoner som utføres i forbindelse med pris- og avansefastsettelse på legemidler. Samarbeidsrutinene og arbeidsfordelingen mellom Rikstrygdeverket og Statens legemiddelkontroll har ikke vært klar nok, og ikke alltid fungert optimalt i saker hvor begge etater forutsettes ha et ansvar. Dette gjelder særlig i saker hvor det både er behov for kompetanse knyttet til praktisering og forvaltning av trygdens bestemmelser og ordninger, og på det farmasøytisk-medisinske feltet knyttet til økonomisk og medisinsk evaluering av legemidler i refusjonssaker. Til tross for at staten i dag er den største betaleren i legemiddelmarkedet, har myndighetene i for liten grad maktet å utnytte sin forhandlingsposisjon til å oppnå gunstige priser på stønadsberettigete legemidler.
Det må understrekes at tilgangen til nye og effektive legemidler er av overordnet betydning for helsevesenet. Myndighetene må likefullt vurdere om samfunnets utgifter til nye legemidler svarer til de medisinske gevinster legemidlet medfører. Dagens saksgang og ansvarsfordeling mellom etatene kan også virke uoversiktlig for produsenter som søker om refusjon. På den annen side må det også tas høyde for at utilfredsstillende resultater knyttet til dagens refusjonsordning også kan skyldes rammevilkårene og de særtrekk som kjennetegner legemiddelmarkedet. Stikkord i den forbindelse er at legen velger bruk av legemiddel (på vegne av pasienten) og at en tredje part (staten) dekker hovedtyngden av utgiftene.
8.1.3.2 Forslag til tiltak
Sosial- og helsedepartementet har inneværende år startet en gjennomgang av organiseringen av den statlige legemiddelforvaltning i samarbeid med sine fagetater. Det er et siktemål for den prosess som er påbegynt at den statlige legemiddelforvaltning må bli bedre og mer slagkraftig på de nevnte saksområder. Sosial- og helsedepartementet vil vektlegge følgende hensyn:
Tilsynsvirksomheten med legers forskrivning av legemidler, legemiddelhåndtering i helsetjenesten, apotek, grossister og farmasøytisk produksjon må fortsatt skje i regi av Statens helsetilsyn. Dette gjelder også for Helsetilsynets faglige rådgivningsfunksjoner. På sikt bør disse funksjoner styrkes. Utviklingen i både helsesektoren og på legemiddelområdet, med krav til effektiv ressursutnyttelse, øket etterspørsel, krav om høy faglig standard, inntog av flere aktører og etablering av nye organisasjonsmodeller (selskapsformer), gjør det nødvendig for myndighetene å videreutvikle tilsynsfunksjonene.
De operative oppgavene Helsetilsynet har innenfor omsetningskjeden for legemidler, slik som konsesjon, avansesystem, apotekstøtteordning, omsetningsavgift og fraktrefusjon, bør vurderes utskilt fra tilsynsfunksjonene med sikte på å kunne rendyrke og forsterke tilsynet og for å unngå dobbeltroller.
Det er videre nødvendig med et tettere og mer forpliktende samarbeid mellom Rikstrygdeverket og Statens legemiddelkontroll for å dra nytte av den samlede fagkompetanse i statsforvaltningen og forhindre etablering av dobbeltfunksjoner. Samtidig må ansvarsforholdene og rapporteringsrutinene gjøres klare.
Statens kompetanse innen legemiddeløkonomi og prisforhandlinger m.v. bør styrkes. Behovet for endringer i den statlige legemiddelforvaltning må således vurderes blant annet i forhold til effektivisering, styrket samarbeid og utvikling av en bedre samhandling innenfor medisinsk effekt, pris og refusjon.
Det legges opp til å fremme forslag om nødvendige organisatoriske endringer med eventuelle budsjettmessige konsekvenser for Stortinget i forbindelse med Revidert nasjonalbudsjett for 1999.
8.2 Koordinering av den statlige veiledningsvirksomheten om riktig legemiddelbruk
En viktig utfordring for myndighetene er å bli bedre til å formidle de faglige forutsetninger knyttet til stønad til legemidler og følge opp om legemidlene brukes riktig faglig og økonomisk. Terapiveiledning og produsentuavhengig legemiddelinformasjon er et viktig satsingsområde. Det er en rivende utvikling på feltet, hvor elektroniske media (inkludert internett) vil spille en stadig viktigere rolle. Det berører styrings- og påvirkningspotensialet for myndighetene i særlig grad, både når det gjelder økonomi og i forhold til helsegevinster.
Mulighetene for helsegevinst hviler på at leger rekvirerer legemidler på rasjonelt grunnlag og at pasienten får nødvendig informasjon og bruker legemidlene i tråd med intensjonene. Terapivalget har avgjørende betydning for kostnadene. Både terapivalg, volum og pris kan påvirkes og optimaliseres. Det arbeides mye både nasjonalt og internasjonalt med utvikling av kunnskapsbaserte retningslinjer for hvordan man gjennom aktiv implementering kan fremme den ønskede endring. Legemiddelindustrien tar i bruk nye virkemidler for å fremme bruk av egne, patenterte legemidler. Her kan nevnes både beslutningsstøttesystemer og økt direkte og indirekte markedsføring av reseptpliktige legemidler direkte til publikum. Dette fører til økt press på leger for valg av legemidler som behandlingsalternativ generelt, og til at etterspørsel mer enn behov og uavhengig kunnskap styrer legemiddelvalg.
Flere offentlige aktører bidrar med informasjon om riktig bruk av legemidler: Norsk legemiddelhåndbok, Institutt for farmakoterapi ved Universitetet i Oslo, de regionale legemiddelinformasjonssentralene, Statens legemiddelkontroll (preparatomtaler, pakningsvedlegg, tidsskriftet «Nytt fra SLK» og terapiverksteder), Helsetilsynet (veiledere og retningslinjer av generell karakter). Dertil kommer Legeforeningens kvalitetssikringsprosjekter, informasjonsarbeidet til Norges Apotekerforening (NAF) og de regionale og lokale legemiddelkomiteene.
Departementet vil løpende vurdere behovet for og samordning av legemiddelinformasjon.
8.3 Norsk tilslutning til Det europeiske legemiddelkontor (EMEA)
Rådsdirektiv 65/65/EØF, som senere er endret en rekke ganger, danner bakgrunnen for det europeiske legemiddelregelverket. Utviklingen siden 1965 har medført en stor grad av harmonisering av vilkårene for innvilgelse av markedsføringstiltatelse for legemidler i landene innenfor EØS-området. Det europeiske legemiddelkontoret (EMEA), som for EU-landene ble innført i 1995, medfører mindre dobbeltarbeid knyttet til at hvert enkelt land gjør sin egen evaluering og godkjenning av nye legemidler etter det samme regelverk. Tidligere var det et tilsvarende nordisk samarbeid, som imidlertid falt bort da Sverige og Finland ble medlemmer av EU. EMEA innebærer også en ensartet godkjenningsprosedyre for legemidler som er utviklet ved hjelp av blant annet bioteknologi.
Markedsføringstillatelse kan oppnås ved en såkalt desentralisert og sentralisert godkjenningsprosedyre. Den desentraliserte prosedyren, gjensidig anerkjennelsesprosedyre, innebærer at en søknad om markedsføringstillatelse som innvilges i en stat, skal anerkjennes også i andre stater. Unntak fra dette utgangspunktet vil være i de tilfeller det er fare for folkehelsen knyttet til legemidlet. Dersom et land er uenig, kan godkjenning klages inn for den sentrale prosedyren. Den sentrale prosedyren skal i tillegg til klagesaker etter den desentraliserte ordningen, i hovedsak gjelde for godkjenning av bioteknologiske og andre viktige, nye høyteknologiske legemidler.
Under den sentrale prosedyren sendes søknadene inn til det sentrale organ, EMEA (European Medicinal Evaluation Agency). Formelt er det EU-kommisjonen som foreslår godkjenning etter anbefaling fra EMEA. For Norge og Island vil de nasjonale legemiddelmyndigheter som i dag er tillagt dette ansvaret, treffe beslutninger nasjonalt. For Norge vil det si Statens legemiddelkontroll. Dersom Norge vil treffe en avgjørelse som avviker fra Kommisjonens, noe Norge vil ha rett til, må saken drøftes i EØS-komiteen for å finne en løsning.
To vitenskapelige komiteer er underlagt EMEA, én komite for legemidler til mennesker og én for legemidler til dyr. Disse komiteene er ansvarlige for evalueringen av legemidlene og består av eksperter fra medlemslandene. Den praktiske gjennomføringen av evalueringene foretas av de to land som blir utpekt som såkalte rapportørland for den enkelte godkjenningssøknad.
EMEA-ordningen innebærer en utvidelse av det tidligere samarbeidet på legemiddelområdet i Europa. Formålet er blant annet å bedre informasjonsutvekslingen mellom landene og sikre en mer enhetlig praktisering av regelverket og en felles overvåkning og registrering av bivirkninger. Systemet medfører en felleseuropeisk godkjenningsmyndighet.
Regjeringen vil legge frem en stortingsproposisjon hvor disse temaene behandles nærmere med henblikk på at EFTA/EØS-landene tilknytter seg samarbeidet i EMEA.