3 Medisinsk-teknologisk kontekst
3.1 Etablert, eksperimentell og utprøvende behandling
Med behandling menes spesifikke tiltak for å fjerne eller redusere symptomer og/eller årsak til sykdom. Behandlingsmetoder innbefatter legemidler, prosedyrer og kirurgiske inngrep. Bruk av diagnostiske metoder er en forutsetning for å iverksette riktige tiltak, men slike metoder kan også være ledd i oppfølgingen av en gitt sykdomstilstand. Digitale verktøy, inkludert kunstig intelligens, vil få økt betydning for både diagnostikk og behandling. I noen sammenhenger vil legemidler, medisinsk utstyr og digitale verktøy inngå sammen som ett tiltak, eksempelvis glukosemålere og insulinpumper.
Det er vanlig å skille mellom etablert behandling og eksperimentell behandling. Etablert behandling er veldokumentert, kommer frem av internasjonale eller nasjonale retningslinjer og representerer dagens praksis (Helsenorge 2023).1Eksperimentell behandling betyr at behandlingen er gjenstand for utprøving gjennom kliniske studier, og at det ikke foreligger tilstrekkelig god dokumentasjon av sikkerhet og effekt. Eksperimentell behandling skal gis innenfor rammen av forskningsprosjekter, med krav om en forskningsprotokoll, forsvarlighet, informasjon, samtykke og rutiner for å overvåke og rapportere bivirkninger.
Begrepet utprøvende behandling er også brukt om behandling som ikke er etablert. Meld. St. 10 (2012–2013) God kvalitet – trygge tjenester— Kvalitet og pasientsikkerhet i helse- og omsorgstjenesten definerer utprøvende behandling som «all behandling der effekt, risiko og bivirkninger ikke er tilstrekkelig dokumentert til at behandlingen kan inngå i det ordinære behandlingstilbudet» (Meld. St. 10 (2012–2013), 105). Det innebærer at utprøvende behandling omfatter både behandling som prøves ut i kliniske studier, og behandling som gis utenfor kliniske studier. Nasjonale føringer som er formulert i Meld. St. 10 (2012–2013) og i Nasjonal veileder for utprøvende behandling (Helsedirektoratet 2021), tilsier at man unntaksvis kan gi utprøvende behandling til pasienter på individuell basis når det er
- Konkludert med at behandlingen er faglig forsvarlig med utgangspunkt i en vurdering av tilgjengelig kunnskap og/eller erfaringer om metodens mulige effekt og sikkerhet
- Gitt tilstrekkelig informasjon om behandlingstilbudet og pasienten aktivt har medvirket i valg av behandlingsmetode
- Etablert protokoller for bruk av metoden
- Utarbeidet rutiner for oppfølging og overvåkning av pasienter som mottar behandlingen
- Etablert rutiner for rapportering av bivirkninger og effekt av behandlingen som systematisk kan sammenstilles for alle pasienter som får behandlingen (Helsedirektoratet 2021, 15).
Grenseflatene mellom etablert, eksperimentell og utprøvende behandling kan være uklare. Det kan for eksempel foreligge god dokumentasjon for bruk av et legemiddel hos én pasientgruppe, men ikke hos en annen. Hos den ene pasientgruppen vil behandlingen da være etablert, mens behandlingen for den andre pasientgruppen er å betrakte som eksperimentell eller utprøvende.
Godkjenningen av bruksområdet for et legemiddel er basert på innsendt dokumentasjon av legemiddelets kvalitet, sikkerhet og effekt. Som hovedregel skal legemidler benyttes i tråd med preparatomtalen (SPC), herunder godkjent indikasjon (bruksområde). I en del sammenhenger brukes legemidler utenfor de rammene preparatomtalene setter. Slik legemiddelbruk utenfor godkjent bruksområde (ofte kalt offlabel-bruk) er vanligvis ansett som utprøvende behandling og er ofte basert på konsensus i fagmiljøet og eventuelt et forskningsgrunnlag. Det finnes eksempler på at den forskningsmessige dokumentasjonen er så god at legemiddelbruk utenfor godkjent bruksområde i praksis er å oppfatte som etablert, selv om legemiddelet formelt ikke har markedsføringstillatelse for det aktuelle bruksområdet.
Kunnskapsgrunnlaget for metoder er dynamisk. Utviklingen av ny kunnskap fortsetter også etter at legemiddelet har fått markedsføringstillatelse eller medisinsk utstyr har oppnådd sertifisering. Det kan være aktuelt å gjennomføre studier for å avklare langtidseffektene av en behandling selv om metoden er godkjent. Følgelig kan man gjennomføre klinisk forskning på etablert behandling for å styrke dokumentasjonsgrunnlaget eller for å sammenlikne den etablerte behandlingen med en ny behandlingsmetode. Ny informasjon om alvorlige bivirkninger kan også føre til at man revurderer bruken av en etablert metode.
3.2 Kunnskap og dokumentasjon
Gode prioriteringsbeslutninger forutsetter kunnskap om metodens forventede effekt. I medisin og helsefag er det vanlig å rangere kvaliteten i kunnskapsgrunnlaget etter kilde, som vist i tabell 1. I en senere metodevurdering vil man også gjøre en vurdering av kvaliteten i dokumentasjonen som foreligger.
Tabell 1. Kunnskapsnivåer i medisinen
Type studie | Kunnskapsnivå | Studiefase |
|---|---|---|
Systematiske oversikter og metaanalyser av randomiserte kontrollerte studier (RCT-er) | 1a | |
Minst én randomisert kontrollert studie | 1b | Fase III |
Minst én godt utformet kontrollert studie uten randomisering | 2a | Fase II |
Minst én annen godt utformet studie uten kontrollgruppe og uten randomisering | 2b | Fase I/II |
Godt utformede deskriptive studier, som sammenliknende studier, korrelasjonsstudier, kasuistikker eller små pasientserier | 3 | |
Rapporter eller oppfatninger fra eksperter, komiteer og/eller klinisk ekspertise hos respekterte autoriteter | 4 |
Tabell 1. Tabellen illustrerer et dominerende syn på medisinsk kunnskap, rangert etter kunnskapsnivåer for dokumentasjonens kvalitet. Øverst finner man systematiske oversikter og metaanalyser av randomiserte kontrollerte studier (RCT-er). Nederst finner man ekspertoppfatninger. Tabellen viser dessuten at utviklingen av et legemiddel – fra fase I til fase III – innebærer en forflytning opp i kunnskapshierarkiet. Fase IV-studier gjennomføres på ulike kunnskapsnivåer og er følgelig ikke ført inn i denne tabellen (Helsedirektoratet 2023a, Statens legemiddelverk 2023a).
3.3 Usikkerhet om effekt og risiko
Utviklingen gjør at sykdomskategorier som tidligere ble oppfattet som ensartede, i økende grad blir mer presist klassifisert i undergrupper basert på biologiske, ofte molekylærgenetiske, karakteristika. Dette medfører at studiedesignet baseres på mindre undergrupper av pasienter som deler biologiske trekk, i stedet for større og biologisk mangfoldige grupper. I noen tilfeller kan det også være aktuelt å gjennomføre single-case-studier med én pasient. Innenfor persontilpasset medisin og sjeldne tilstander kan det være utfordrende å gjennomføre studier av god vitenskapelig kvalitet og statistisk styrke.
3.4 Persontilpasset medisin
Regjeringen lanserte i 2023 Nasjonal strategi for persontilpasset medisin 2023–2030, hvor persontilpasset medisin (også kalt presisjonsmedisin) defineres som «økt grad av tilpasning til den enkeltes biologi, som ideelt sett øker sannsynligheten for at valgte terapeutiske tiltak gir effekt og ledsages av færre bivirkninger» (Regjeringen 2023, 13).
Målet er å skreddersy behandlingen til den enkeltes biologiske sykdomstilfelle gjennom presis diagnostikk og behandling. Sentralt i persontilpasset medisin står molekylærbiologiske undersøkelser av pasientens genmateriale eller molekylære egenskaper ved selve sykdommen. Tilstrekkelig kapasitet og kompetanse i analyser av gener og molekylære egenskaper ved biologiske prosesser er avgjørende for å identifisere distinkte undergrupper og individer.
Ganske ofte vil persontilpasset medisin innebære en tilnærming hvor man konsentrerer seg om undergrupper av pasienter med like egenskaper. Det tar tid å samle et stort nok antall mennesker i en klinisk studie, og internasjonalt samarbeid for å rekruttere pasienter er derfor viktig. Imidlertid vil dokumentasjonen i slike studier kunne ha lavere kvalitet enn det man normalt forventer for større pasientgrupper, noe som kan føre til usikkerhet om effekten og risikoen ved behandlingen.
Det finnes eksempler hvor man bruker metoder for individualiserte behandlingsopplegg der den enkelte pasienten representerer enheten som blir gjenstand for en klinisk studie, kalt single-case design studies, som blant annet inkluderer «n-of-1 trials». Omfanget av slike studier er begrenset, men det er grunn til å tro at forekomsten vil øke med videreutviklingen av persontilpasset medisin. Kunnskap om et forventet forløp og ulike mål (biomarkører) for sykdomsaktivitet vil være viktig for å vurdere om det man observerer i denne ene pasienten, representerer en reell behandlingseffekt. I den grad det foreligger pålitelig dokumentasjon om effekten, vil man kunne foreta en vurdering av forholdet mellom behandlingens kostnad og dens forventede effekt.
Utviklingen i persontilpasset medisin innebærer at det vil være behov for mer dynamiske systemer for metodevurdering og prioritering (Regier et al. 2022).
3.5 Sjeldne sykdommer
En sjelden tilstand er definert som en helsetilstand med lav prevalens, det vil si at den veiledende forekomsten er færre enn 5 av 10 000 innbyggere (HOD 2021a). I Norge har vi ingen egen definisjon av «ultrasjeldne» diagnoser. Internasjonalt er det vanlig å definere en ultrasjelden sykdom som en sykdom som færre enn 1 av 50 000 har.
Sjeldne sykdommer er temmelig vanlige på befolkningsnivå. Nasjonalt kompetansesenter for sjeldne sykdommer anslår at det finnes om lag 7000 ulike sjeldne sykdommer i Norge, og at 3,5–5,9 prosent av befolkningen har en sjelden diagnose. 80 prosent av disse sjeldne sykdommene er arvelige (Helsenorge 2023).
Utviklingen av legemidler frem mot markedsføringstillatelse følger i prinsippet samme trinnvise vei (jf. tabell 1), uavhengig av forekomst. På sjeldenfeltet har det vært en utfordring å ha tilstrekkelig forskningsaktivitet og utvikling av behandlinger, blant annet på grunn av de økonomiske ulempene knyttet til utviklingen av legemidler for svært små pasientgrupper. I både EU og USA er det utviklet egne virkemidler som skal stimulere til utvikling av legemidler for sjeldne sykdommer (såkalt orphandesignation). Regelverket er del av EØS-avtalen og gjelder som forskrift, jf. forskrift 18. desember 2009 nr. 1839 om legemidler til mennesker (legemiddelforskriften) § 15-7 (Norsk legemiddelhåndbok 2023). Det finnes spesielle regulatoriske godkjenningsordninger som også kan være aktuelle for legemidler for sjeldne sykdommer. Såkalt betinget markedsføringstillatelse kan være aktuelt for legemidler som fyller et udekket behov for behandling av alvorlig sykdom, selv om data som normalt kreves for godkjenning, mangler. Det er en forutsetning at fordelen ved rask tilgang oppveier risikoen ved mangel på kliniske data.
Noen få legemidler godkjennes under eksepsjonelle omstendigheter, eksempelvis når det er svært få pasienter, eller når det vil være uetisk å gjennomføre ytterligere kliniske studier. Begge disse godkjenningsordningene er ledsaget av spesifikke oppfølgingskrav om fortsatt kunnskapsgenerering.
Metoder rettet mot særskilt små pasientgrupper med svært alvorlig tilstand vurderes i dag i de etablerte beslutningssystemene på gruppenivå, dvs. i spesialisthelsetjenesten innenfor Nye metoder og under systemet for forhåndsgodkjent refusjon (§ 2). Ved metodevurdering og beslutning om innføring av slike legemidler kan det aksepteres et lavere krav til dokumentasjon og en høyere ressursbruk enn det som normalt aksepteres (Meld. St. 34 (2015–2016), 13). I 2017 ble det etablert tre veiledende kriterier som alle må være oppfylt for at metoder kan kvalifisere til ordningen for særskilt små pasientgrupper med svært alvorlig sykdom. Disse er som følger:
- Særskilt liten pasientgruppe: a) Mindre enn ca 1 pasient per 100 000 innbyggere på verdensbasis per legemiddel b) Mindre enn cirka 50 pasienter i Norge per legemiddel.
- Svært alvorlig tilstand: Alvorlighetsgrad målt ved absolutt prognosetap tilsvarer minimum cirka 30 tapte gode leveår.
- Stor forventet nytte av legemiddel: Forventet nytte av den aktuelle behandlingen er betydelig og minimum cirka 2 vunnet gode leveår sammenlignet med standard behandling.
Alle disse tre veiledende kriteriene skal som et utgangspunkt være oppfylt for at et legemiddel skal kunne vurderes iht. legemiddelforskriften § 14-5 tredje ledd (Statens legemiddelverk 2023b).
Det er få (kun en håndfull) legemidler som er vurdert å oppfylle kriteriene for denne ordningen siden den ble innført. Kravene til kunnskap og dokumentasjon for prioritering som er differensiert basert på sykdommers sjeldenhet, er også beskrevet i kapittelet om prioritering av offlabel- og offlicence-bruk (kapittel 3.8) og er etablert for å ivareta en rettferdig prioritering for disse pasientene sammenliknet med pasienter med vanligere diagnoser.
Man forventer at større presisjon ved diagnostiske metoder og nye, målrettede behandlingsmetoder vil åpne for økt differensiering av sykdommer. Antallet sjeldne og svært sjeldne undergrupper innenfor allerede kjente sykdomsgrupper vil dessuten øke med den molekylærteknologiske og biologiske utviklingen. I tillegg får man stadig mer detaljert kunnskap om de molekylære årsakene (driverne) for sykdomsutviklingen. Slik kunnskap om molekylære sykdomsdrivere gir ofte muligheten til å utvikle nye målrettede legemidler. Eksempelvis ble det i USA i perioden 2011–2020 gitt markedsføringstillatelse årlig for 40 nye virkestoff (New MolecularEntities), som er en økning sammenlignet med perioden 2001–2010 (PhRMA 2021).
Å fremlegge nødvendig dokumentasjon av kvalitet, sikkerhet og effekt for å oppnå markedsføringstillatelse er mulig selv ved svært sjeldne sykdomstilstander. Et illustrerende eksempel er den sjeldne sykdomsgruppen arvelig retinal dystrofi (se boks 1).
Boks 1. Retinal dystrofi som et eksempel på vurdering ved sjeldenhet
Retinal dystrofi er en sykdom som gir gradvis økende synstap. Det er estimert at det finnes nærmere 300 ulike gener hvor en mutasjon kan forårsake en tilstand som tilhører denne sykdomsgruppen. For en svært liten pasientgruppe med genfeil i ett av disse 300 genene, RPE65, er det utviklet et legemiddel som kan bedre synsevnen. Dette legemiddelet fikk markedsføringstillatelse basert på en innledende fase I-studie og en oppfølgende randomisert kontrollert fase III-studie med under 50 deltagere. Kunnskapsgrunnlaget for å beregne kostnadseffektiviteten av denne behandlingen er ennå svært usikkert, men det vil bli sikrere med lengre oppfølgingstid. Legemiddelet ble godkjent av Beslutningsforum for nye metoder, med forbehold om senere endringer, avhengig av oppfølgende data. I utgangspunktet gir man behandlingen bare én gang, og det hersker i skrivende stund fortsatt usikkerhet om hvordan effekten (og eventuelle bivirkninger) vil utvikle seg over tid (Statens legemiddelverk 2021).
De fleste sjeldne tilstander som er kjent i dag, har en forekomst som gjør det mulig å gjennomføre kliniske studier på fase I-nivå med mer enn én deltaker. Det er imidlertid publisert minst ett tilfelle der man i dag bare kjenner til én pasient med den aktuelle tilstanden, og hvor det er utviklet et legemiddel for denne (Kim et al. 2019).
Det stilles store krav til dokumentasjon av sikkerhet før et legemiddel gis til mennesker, uavhengig av hvor mange pasienter man planlegger å inkludere i studieprogrammet. Slik preklinisk dokumentasjon forelå også i eksempelet ovenfor; typen legemiddel (antisense-oligonukleotid) var kjent, og det var liten sannsynlighet for at legemiddelet ville kunne gi irreversibel og alvorlig skade. Legemidler av denne typen reparerer ikke genfeilen, men kan påvirke proteinsyntesen i en ønsket retning. Etterlevelse av god forskningsetikk, i særdeleshet krav til frivillig, informert samtykke og ivaretakelse av integritet og personvern, blir imidlertid særlig krevende i situasjoner som den som er beskrevet ovenfor. Det vil kunne være en tett kobling mellom pasient, utviklere og patenteiere, og det kan oppstå urealistiske forventninger til legemiddelets nytte-/risikoforhold. Videre vil det kunne oppstå uklarheter eller uenighet om hvilke resultatmål som er nødvendige eller tilstrekkelige for å videreføre behandlingen, som i dette tilfellet vil kunne være livsvarig.
Det kan være krevende å gjennomføre metodevurderinger for behandlingen av sjeldne sykdommer, fordi kunnskapsgrunnlaget er usikkert. Fra et prioriteringsståsted synes hovedproblemet i første rekke å være kostnadene. Den videre teknologiske utviklingen vil vise om kostnad–effekt-forholdet kommer på et nivå som gjør det mulig å prioritere metoder i situasjoner med målrettede legemidler skreddersydd for svært sjeldne sykdommer. Den amerikanske etaten U.S. Food and Drug Administration (FDA) har arbeidet med å etablere et regelverk som skal ivareta problemstillinger om sjeldne sykdommer (FDA 2021).
3.6 Undergrupper i studiepopulasjoner
Det vil alltid være variasjon blant pasienter som deltar i en klinisk studie. Følgelig er det vanlig å undersøke i hvilken grad effekten av studieintervensjonen (f.eks. behandling med et nytt legemiddel) varierer i definerte undergrupper. Disse undergruppene bør være basert på karakteristika som man med rimelighet tror har betydning for effekten av behandlingen, som kjønn, alder, sykdomsutbredelse, omfang av tidligere behandling, kliniske funn og definerte biomarkører (biologiske egenskaper ved pasienten eller sykdommen). Slike karakteristika for undergrupper definerer man oftest i en tidlig studiefase (fase I eller II). Fase III-studiedata er imidlertid oftest nødvendig for å skille mellom underliggende faktorer som kan si noe om forløpet (prognose), og forskjeller som skyldes ulik effekt av selve intervensjonen.
3.7 Utviklingen av nye legemidler
Legemidler kan utvikles i laboratorium og eventuelt ved bruk av forsøksdyr. Sikkerhet og effekt for legemiddelet blir undersøkt i kliniske studier på mennesker. Dette skjer i ulike studiefaser, fra fase I inntil fase III, der målet er å oppnå regulatorisk godkjenning (markedsføringstillatelse, MT) fra legemiddelmyndighetene på et definert bruksområde (indikasjon).
I fase I av utprøvingen blir sikkerhetsprofilen (dvs. bivirkninger) ved bruk dokumentert. I tillegg blir legemiddelets egenskaper og sannsynlighet for effekt undersøkt med en tilnærming som gir en pekepinn om effekt. I fase II blir effekt og bivirkninger dokumentert med data fra flere pasienter, lengre observasjonstid og sikrere endepunkter. Som regel foregår også fase II uten noen kontrollgruppe. I fase III inkluderer man en kontrollgruppe som får etablert behandling, som regel basert på randomisering. Dette kalles en randomisert klinisk studie. Antallet pasienter blir dessuten ytterligere økt. Slik blir det, i fase III, mulig å dokumentere effektstørrelsen for det nye legemiddelet, målt med et ønsket endepunkt og sammenliknet med etablert behandling.
Etter at man har oppnådd markedsføringstillatelse på en gitt indikasjon, blir det av og til gjennomført såkalte fase IV-studier hvor formålet er å innhente ytterligere informasjon om sikkerhet og effekt i en bredere pasientpopulasjon. Disse studiene er utformet for å fange opp eventuelle sjeldne bivirkninger og langtidseffekter, eller undersøke nye kliniske eller helseøkonomiske utfallsmål (Statens legemiddelverk 2023a). Det er i tillegg ganske vanlig at et legemiddel som har oppnådd markedsføringstillatelse på én indikasjon, blir undersøkt i kliniske studier for bruk på andre indikasjoner, eller over en lengre periode. Slike studier kan ha så vel fase II- som fase III-design. Figur 1 og 2 nedenfor gir en oversikt over tilgang til nye metoder gjennom henholdsvis de regionale helseforetakene (RHF-ene) og folketrygden.
Figur 1. Tilgang til nye sykehusfinansierte legemidler
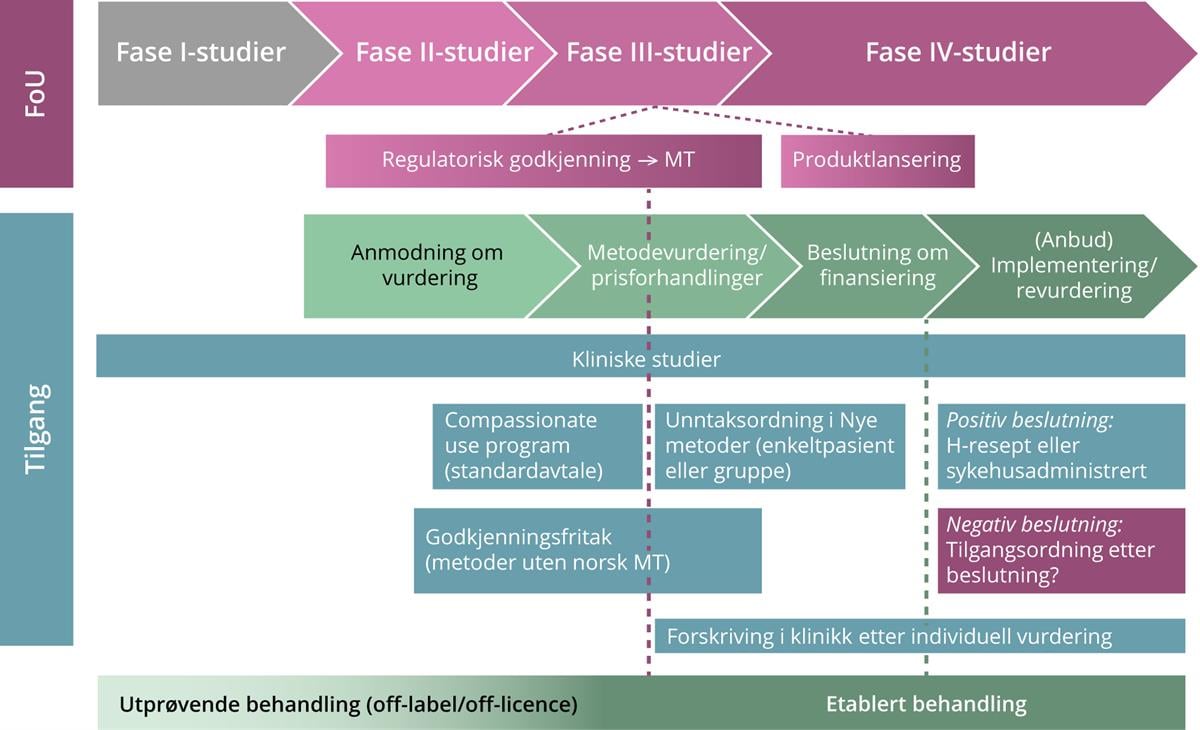
Figur 1. Prosessen frem mot offentlig tilgang til nye metoder gjennom de regionale helseforetakene.
Figur 2. Tilgang til nye folketrygdfinansierte legemidler
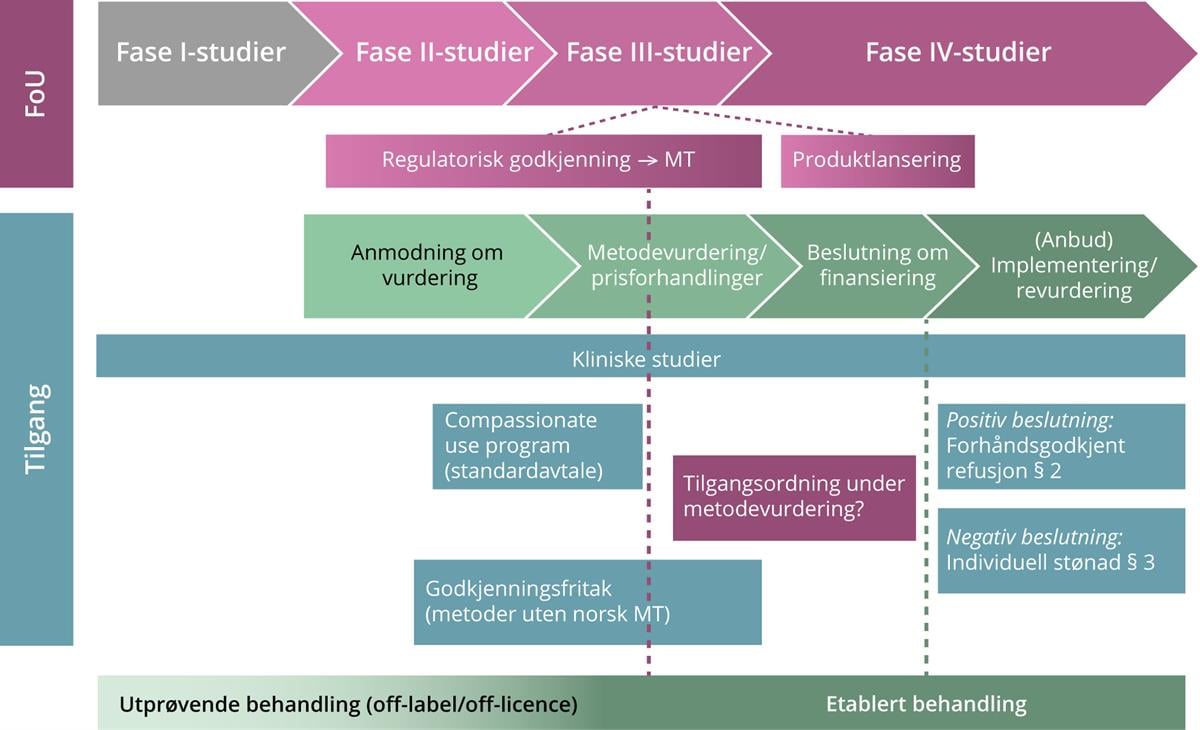
Figur 2. Prosessen frem mot offentlig tilgang til nye metoder gjennom folketrygden (blåreseptordningen).
Alle nye og innovative legemidler skal vurderes i sentral prosedyre. Legemidler som godkjennes i sentral prosedyre, får markedsføringstillatelse i alle EU/EØS-land. Får et legemiddel innvilget regulatorisk godkjenning, innebærer det at det europeiske legemiddelbyrået European Medicines Agency (EMA) har vurdert at legemiddelet har et dokumentert positivt forhold mellom forventet nytte og risiko. Den regulatoriske godkjenningen innebærer imidlertid ikke at legemiddelet skal finansieres av den offentlige helsetjenesten. Slike beslutninger tas nasjonalt, i mange land basert på en metodevurdering (Health Technology Assessment (HTA)) som beregner helsegevinster og kostnader ved å ta i bruk den nye metoden, sammenliknet med eksisterende standard behandling for en pasientgruppe. I Norge gjøres metodevurderinger gjennom Nye metoder for sykehusfinansierte metoder, og gjennom blåreseptordningen for legemidler finansiert av folketrygden.
Vurderinger i Nye metoder og blåreseptordningen forutsetter at det foreligger troverdige data for sikkerhet og effekt både for den aktuelle pasientgruppen (case) og for en mest mulig lik kontrollgruppe med standardbehandling. I praksis innebærer denne forutsetningen at det i de fleste tilfellene må foreligge dokumentasjon på nivå 1b eller bedre (jf. tabell 1), men det er ikke et krav om at det må foreligge slik dokumentasjon for å gjøre en metodevurdering. Randomiserte kliniske studier representerer gullstandarden, men de siste 10–15 årene har man i økende grad sett studier med bare én gruppe (enarmede studier (single arm-studier)), spesielt på kreftområdet. Nivå 1b foreligger vanligvis for legemidler som har fått markedsføringstillatelse, og langt sjeldnere for andre metodekategorier i helsetjenesten (f.eks. tester og prosedyrer).
3.8 Bruk av legemidler utenfor godkjent indikasjon
Det er en åpning for at leger kan rekvirere legemidler utenfor godkjent indikasjon (offlabel) eller legemidler uten norsk markedsføringstillatelse (offlicence). Særlig for metoder hvor det ikke foreligger markedsføringstillatelse verken på europeisk eller nasjonalt nivå, vil dokumentasjonen kunne være mangelfull, eventuelt at markedsføringstillatelse ikke er søkt. Både offlabel- og offlicence-bruk anses å være utprøvende behandling og stiller krav til registrering, til at pasienten er informert, og til at behandleren er spesielt årvåken for eventuelle bivirkninger. De regionale helseforetakene har laget et Rammeverk for legemiddelbehandling utenfor godkjent indikasjon i spesialisthelsetjenesten (RHF-ene 2022) og Retningslinjer for bruk av nye legemidler før markedsføringstillatelse (Nye metoder 2023).
I sine kommentarer til forskrift 28. juni 2007 nr. 814 om stønad til dekning av utgifter til viktige legemidler mv. (blåreseptforskriften) § 3 fjerde ledd oppgir Helsedirektoratet krav til dokumentasjon for refusjon ved så vel offlabel- som off licence-bruk:
Som hovedregel kreves det at det foreligger randomiserte og kontrollerte studier av en viss størrelse. Der aktuell studie viser særlig god og klinisk relevant effekt, kan Helfo senke kravet til studiepopulasjonens størrelse. For mindre pasientpopulasjoner (inntil 400 personer i Norge) kan observasjonsstudier i form av kohort-studier eller kasus-kontroll-studier unntaksvis godtas. Studiene må vise til god og klinisk relevant effekt. Kravet anses som oppfylt hvis den aktuelle legemiddelbehandlingen er anbefalt i nasjonale faglige retningslinjer utarbeidet eller godkjent av Helsedirektoratet (Helsedirektoratet 2023b, 18–19).
Dette innebærer at dokumentasjonsgrunnlaget i enkelte saker ikke behøver å være på høyere kunnskapsnivå enn 3 (se tabell 1). Helsedirektoratet angir også eksempler på dokumentasjon som ikke vektlegges:
Som følge av krav til vitenskapelig godt dokumentert effekt vektlegges ikke
1. klinisk erfaring eller opplevd effekt hos den enkelte pasienten.
2. retningslinjer fra andre land og norske retningslinjer utarbeidet av ulike spesialistmiljøer (unntak barn og gravide).
3. sannsynlig effekt ut fra kjemisk struktur eller effekt på beslektede diagnoser/symptomer (Helsedirektoratet 2023b, 19).
For offlicence-behandling er det etablert en nasjonal avtaleordning som skal sikre lik tilgang for alle aktuelle pasienter i avtaleperioden. Offlabel-bruk er regulert i Rammeverk for legemiddelbehandling utenfor godkjent indikasjon i spesialisthelsetjenesten (RHF-ene 2022). Rammeverket angir dokumentasjonskrav som likner de i blåreseptordningen, med differensiering etter pasientgruppens størrelse.
Fase-II studier, eventuelt retrospektive pasientserier/registerdata, kan være tilstrekkelig, enten for små pasientgrupper der fase-III studier ikke er gjennomførbare, eller for større pasientgrupper hvor dokumentasjonen foreløpig er utilstrekkelig for markedsføringstillatelse. Et minimumskrav er at dokumentasjonen må foreligge i form av en fullverdig originalartikkel i et anerkjent fagfellevurdert fagtidsskrift. For svært små pasientgrupper (insidens færre enn 5 pasienter per år i Norge) kan kasuistikk være tilstrekkelig, særlig dersom gruppen er mulig å identifisere med en prediktiv markør som sannsynliggjør effekt («persontilpasset behandling») (RHF-ene 2022, 12).
På samme vis som ved praktisering av individuell stønad i blåreseptordningen kan kunnskapsgrunnlaget være av lav kvalitet (nivå 3). Videre er det presisert at ordningen er ment å skulle gi konsistent prioritering, uavhengig av populasjonsstørrelse: «Kostnaden ved behandlingen, relativt til helsegevinsten, må etter beste skjønn kunne vurderes til å være i samme størrelsesorden som kostnad–nytte-nivåer i saker som er besluttet i Beslutningsforum» (RHF-ene 2022, 12–13).